Dutch sequels of Tofutown saga: Thou Shall Not Touch Dairy Names!
Posted: July 25, 2024 Filed under: Advertising, alternative protein, Authors, Enforcement, Food, Information Comments Off on Dutch sequels of Tofutown saga: Thou Shall Not Touch Dairy Names! On 23 July 2024 a Dutch Court ruled in summary relief proceedings that Upfield cannot use the name “roombeter” for a plant-based alternative for butter, as this is in violation of the Regulation establishing a common organization of the markets for agricultural products (“COM Regulation”). You should know that “roombeter” translates in English as a composition of “cream” and “better”, whereas “cream” is a reserved designation under the COM Regulation that can only be used for dairy products. Furthermore, “beter” is close to “boter”, being the Dutch designation for butter.
On 23 July 2024 a Dutch Court ruled in summary relief proceedings that Upfield cannot use the name “roombeter” for a plant-based alternative for butter, as this is in violation of the Regulation establishing a common organization of the markets for agricultural products (“COM Regulation”). You should know that “roombeter” translates in English as a composition of “cream” and “better”, whereas “cream” is a reserved designation under the COM Regulation that can only be used for dairy products. Furthermore, “beter” is close to “boter”, being the Dutch designation for butter.
Facts of the case at hand
In the case at hand, Upfield markets a plant-based alternative for butter under the brand BLUE BAND and the product name ROOMBETER. The packaging of the product furthermore states “100 % plant-based alternative for butter” and “81 % less climate impact than butter”. The packaging itself consists of golden coloured paper that is also used for conventional butter in the Netherlands and it displays a curl of butter as shown below. The Dutch Dairy Association opposed the use of the product name ROOMBETER, as it is considered this a violation of the COM Regulation, as explained below.
Case before Dutch Advertisement Code Committee
Prior to this legal procedure , the Dutch Dairy Association had submitted a complaint regarding this product before the Dutch Advertisement Code Committee. This self-regulatory body ruled on March 21 last that the presentation of the product was misleading, since it could be understood to contain butter. The “e” in “beter” could be confused for an “o”, resulting in “boter”, which is Dutch for butter. And furthermore the golden coloured packaging added to the misleading character of the product. The topic of violation of the COM Regulation was left to civil law proceedings, as it exceeded the competence of the Committee.
Applicable legislation
Article 78.2 of the COM Regulation states that the definitions, designations and sales descriptions provided for in its Annex VII may be used in the Union only for the marketing of a product that conforms to the corresponding requirements laid down in that Annex. Annex VII contains, amongst other things, a product definition for milk and a list of milk products. It furthermore states that these designations may not be used for any other product than milk and milk products. The purpose of this provision is to protect dairy names from being used for non-dairy products.
Tofutown
You may recall that in its Tofutown decision back in 2017, the ECJ formulated a very strict prohibition of the use of diary names for non-dairy products (check out our blog on this case here). As a result of that prohibition, the use of the designation “Tofubutter” for a tofu-based product was in violation of the COM Regulation. As a general rule, the ECJ precluded the term ‘milk’ and the designations reserved by the COM Regulation exclusively for milk products from being used to designate a purely plant based product in marketing or advertising. This even applies if those terms are expanded upon by clarifying or descriptive terms indicating the plant origin of the product at issue
Arguments in favour of ROOMBETER
Upfield had argued it did not market its product under the designation ROOMBETER but under the designation BLUE BAND ROOMBETER. The brand BLUE BAND has been used for more than 100 years for margarine, so it is obvious for the consumer this is a plant-based product This is even strengthened by the Dutch translation of the designations “100 % plant-based alternative for butter” and “81 % less climate impact than butter”. So the name ROOMBETER does not designate, imply or suggest it is about a dairy product.
Court decision
The Court did not eat it. Instead, it very strictly applied the Tofutown doctrine, stating that a reserved designation under the COM Regulation cannot be used for a plant-based product. It went on to explain that if it is prohibited to use the designation “tofubutter” for a plant-based product, as it contains the reserved designation “butter”, for sure it is prohibited to use the designation “roombeter” for a plant-based product, as it contains another reserved designation under the COM Regulation. Also, the element “beter” (“better” in English) can hardly be perceived as a clarifying or descriptive term, as it does not refer (contrary to “tofu”) to a plant-based origin. In fact, its reference to plant-based origins can only be understood by those consumers who know the “skip the cow” ad or who further study the packaging of this product.
Upfield was therefore ordered to stop using the designation ROOMBETER within three months after the date of the legal decision.
Consequences of this decision
Should the conclusion of this decision be that any reference to dairy products should be meticulously avoided when marketing plant-based dairy replacements? This seems a very hard task, as manufacturers of these replacement products will want to indicate how their products can be used. Happily, this is not the case. It is still permitted to mention that your plant-based product is for instance a “yoghurt variation”, as this is perceived as a product explanation rather than a product designation. This is not in violation of the Tofutown doctrine and in line with a 2019 Dutch Supreme Court decision relating to a soy-based product marketed by Alpro. Advertising plant-based dairy alternatives nevertheless remains a delicate balancing act.
European harmonization probiotic claims possible? Here’s the latest.
Posted: April 24, 2024 Filed under: Advertising, Authors, Food, Food Supplements, Health claims, Information, Nutrition claims Comments Off on European harmonization probiotic claims possible? Here’s the latest. The International Probiotics Association Europe (IPA Europe) is calling for harmonized use of the claim ‘probiotics’ in the EU. Aforementioned term is generally considered an unauthorized health claim under the EU Claims Regulation. Nevertheless, an increasing number of EU member states allows the use of this claim under certain conditions. This blogpost dives into the regulatory status of probiotic claims in different EU member states and the latest developments in this area.
The International Probiotics Association Europe (IPA Europe) is calling for harmonized use of the claim ‘probiotics’ in the EU. Aforementioned term is generally considered an unauthorized health claim under the EU Claims Regulation. Nevertheless, an increasing number of EU member states allows the use of this claim under certain conditions. This blogpost dives into the regulatory status of probiotic claims in different EU member states and the latest developments in this area.
When entering the keyword ‘probiotic’ in the EU Health Claims Register, one is confronted with over 100 rejected health claims. Over the past years, this has led to the question whether the term ‘probiotic’ should be allowed under certain conditions, and has resulted into divergent policies in different EU member states. To protect both the food industry (against unfair competition) and the consumer (against misleading information), these divergent policies are reason for IPA Europe to call for a harmonized framework.
Czech Republic, Northern Ireland and France
Although EU member states generally consider ‘probiotics’ to be an unauthorized health claim under the EU Claims Regulation, some EU member states take a different approach. ‘Probiotics’ is for example considered a nutrition claim in the Czech Republic. Such claim is allowed if the conditions set forth in the EU Claims Regulation are met. This means, amongst others, that the good bacteria in question are present in the food in an amount that, according to generally accepted scientific evidence, causes the claimed beneficial effect. By contrast, in France and Northern Ireland, the term ‘probiotics’ can be used as a general, non-specific health claim that is allowed in combination with the authorized health claim “live cultures in yoghurt or fermented milk improve lactose digestion of the product in individuals who have difficulty digesting lactose”.
Italy
Italy takes the view that the term ‘probiotics’ does not meet the definition of a health claim and therefore falls outside the scope of the EU Claims Regulation. Italy supports this view by EFSA’s conclusion that probiotic colonization in the intestinal flora (without further specification of bacterial species or strains) is insufficient evidence to substantiate a beneficial effect on human health. In other words, no link between probiotics and health can be demonstrated, for which reason ‘probiotics’ cannot be considered a health claim. Aforementioned reasoning is however not a free pass to unconditionally use the term ‘probiotics’ in Italy. Instead, the following conditions must be met if and when using this term: (i) safety for human consumption, (ii) a history of use for the benefit of the intestinal flora, and (iii) presence of the relevant bacteria in the food in live form and in an adequate quantity until end of shelf life.
Denmark and Spain
In Denmark, the term ‘probiotics’ can be used based on a different legal ground, namely as a mandatory category designation under the EU Food Supplements Directive. In France, such is also possible. Indeed, Article 6(3)(a) of the EU Food Supplements Directive requires that the labeling of the food supplement shall bear “the names of the categories of nutrients or substances that characterize the product or an indication of the nature of those nutrients or substances”. In Spain, the claim ‘probiotics’ is allowed thanks to the principle of mutual recognition. Based on this principle, a product lawfully marketed in one EU member state must also be accepted on the market of another EU member state. Since probiotic claims are therefore allowed on the Spanish market for products from other EU member states, banning the term ‘probiotics’ at national level would discriminate against national producers. Therefore, both food products produced within Spain and those produced outside of the country can bear the claim ‘probiotics’.
Netherlands
In the Netherlands – just like in Denmark and France – the term ‘probiotics’ can be used as a category designation for food supplements. This Dutch practice is laid down in the Guideline Document on the EU Claims Regulation by the Dutch Health Advertising Knowledge and Advice Council (in Dutch: Keuringsraad). An earlier version of the Nutrition and Health Claims Manual by the Dutch Food Safety Authority (in Dutch: NVWA Handboek Voedings- en Gezondheidsclaims) also explicitly mentioned this possibility. Such is now longer mentioned in the latest version of the Nutrition and Health Claims Manual, which can be explained by the fact that ‘probiotics’ as a category designation for food supplements is not a nutrition or health claim.
Although claims that further elaborate on the health effect of probiotics (sporadically) occur on the Dutch (online) market, these are not allowed. Whether the expression “increases the good bacteria in the intestinal flora” (without using the term ‘probiotics’) is acceptable in the Netherlands, is yet unclear. Yakult uses this expression to advertise its fermented milk drink. The Dutch Health Advertising Knowledge and Advice Council would not allow this expression in the context of its preventive supervision in the context of food supplements, because this expression in effect creates a link between the food product and health. If the NVWA has however a different opinion on this and does consider aforementioned expression possible, then other food businesses can take advantage of this too. The call for harmonized uses of the term ‘probiotics’ by IPA Europe, about which more below, can possibly contribute to the acceptance of such expression at national level.
”Probiotics’ not a health claim”
As demonstrated above, various EU member states have introduced national rules on the use of the term ‘probiotics’. As a result, IPA Europe believes that the European Commission’s position that ‘probiotics’ implies a health benefit and is therefore a(n unauthorized) health claim no longer holds water. Instead, IPA Europe advocates qualifying the claim ‘contains probiotics’ as a nutrition claim, just like ‘contains vitamins and minerals’ and ‘contains fiber’. Aforementioned substances may have beneficial nutritional properties, but no specific health benefit is claimed. To strengthen its argument that ‘probiotics’ is not a health claim, IPA Europe explains that the term is not sufficiently precise to substantiate the claimed health benefit under reference to an EFSA guidance document published in 2016.
Call for harmonized use of the term ‘probiotics’
According to IPA Europe, ‘probiotics’ is therefore not a health claim and does not require authorization under the EU Claims Regulation. At the same time, it emphasizes the need for clear rules on the use of the claim as this contributes to a fair competitive environment for food businesses and helps consumers to make informed choices. In December 2023, IPA Europe therefore called for a clearly defined framework for the use of the term ‘probiotics’ in the EU. IPA Europe recommends the following four criteria for consistent use thereof:
- characterization of the species level and identification of at strain level;
- the probiotic strain must be safe for the intended use, e.g. based on the QPS list;
- the probiotic status should be scientifically documented; and
- the probiotic strains must be alive in the product and in a sufficient amount up to the end of shelf-life.
Final comment
The EU knows a fragmented regulatory landscape when it comes to the use of the term ‘probiotics’. Despite IPA Europe’s efforts for a harmonized approach throughout the EU, we will need European legislation or a ruling from the European Court of Justice to have the same rules in all EU member states. For now, the term ‘probiotics’ is (fortunately) not completely banned in the Netherlands and neither in quite a few other EU Member States.
This blogpost has also been published in Dutch at VMT.nl.
New Genomic Techniques: threat or opportunity?
Posted: October 23, 2023 Filed under: Authors, Food, New Genomic Techniques, organic Comments Off on New Genomic Techniques: threat or opportunity?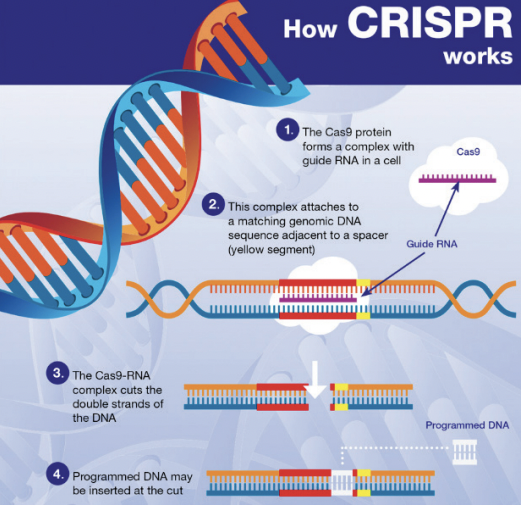 Genetically modified organisms (GMOs) do not generally receive a warm welcome from the average EU citizen. Possibly this is a case of ‘unknown makes unloved’. But GMOs can also bring positive things, for example, in the field of plant breeding. This summer, the European Commission published a proposal for an NGT Regulation. What exactly does this proposal entail and what are the expected consequences in practice? And how was this proposal received by the European Parliament in its draft report of 16 October last in the first reading of the legislative procedure?
Genetically modified organisms (GMOs) do not generally receive a warm welcome from the average EU citizen. Possibly this is a case of ‘unknown makes unloved’. But GMOs can also bring positive things, for example, in the field of plant breeding. This summer, the European Commission published a proposal for an NGT Regulation. What exactly does this proposal entail and what are the expected consequences in practice? And how was this proposal received by the European Parliament in its draft report of 16 October last in the first reading of the legislative procedure?
Potential benefits GMOs and reluctance
GMOs can help develop plants that are more drought tolerant or less susceptible to certain fungi. This reduces the need for pesticides in cultivation, precisely one of the objectives of the EU Farm-to-Fork strategy that the Commission published at the beginning of its mandate in May 2020. At the same time, in the Netherlands the proposal for the NGT Regulation prompted Odin, Demeter, Ekoplaza and Greenpeace, among others, to start a petition calling for “Keep our food genetically modified free.” (in Dutch: Houd ons voedsel gentech vrij). At the time of writing this blogpost, the petition counts 47.709 signatories.
Scope of application NGT Regulation
The intended Regulation applies to NGT plants and to NGT products, meaning food and feed containing or consisting of or produced from NGT plants and other products containing or consisting of such plants. NGT plants are obtained from the following two genomic techniques or a combination thereof:
- Targeted mutagenesis: this is a technique that results in changes to the DNA sequence at precise locations in an organism’s genome;
- Cisgenesis: this is a technique that results in the insertion, into the genome of an organism, of genetic material already present in the total genetic information of that organism or another taxonomic species with which it can be crossed.
In essence, NGTs are genomic techniques such as CRISPR-Cas9. These techniques result in more targeted genomic modifications than older genomic techniques, which often involve the introduction of heterologous genetic material. The European Commission therefore recognizes that any risks associated with the use of NGTs are lower than those associated with older genetic techniques. This is what the risk assessment in the draft Regulation specifically addresses.
Two specific regulatory regimes for category 1 and category 2 NGT plants
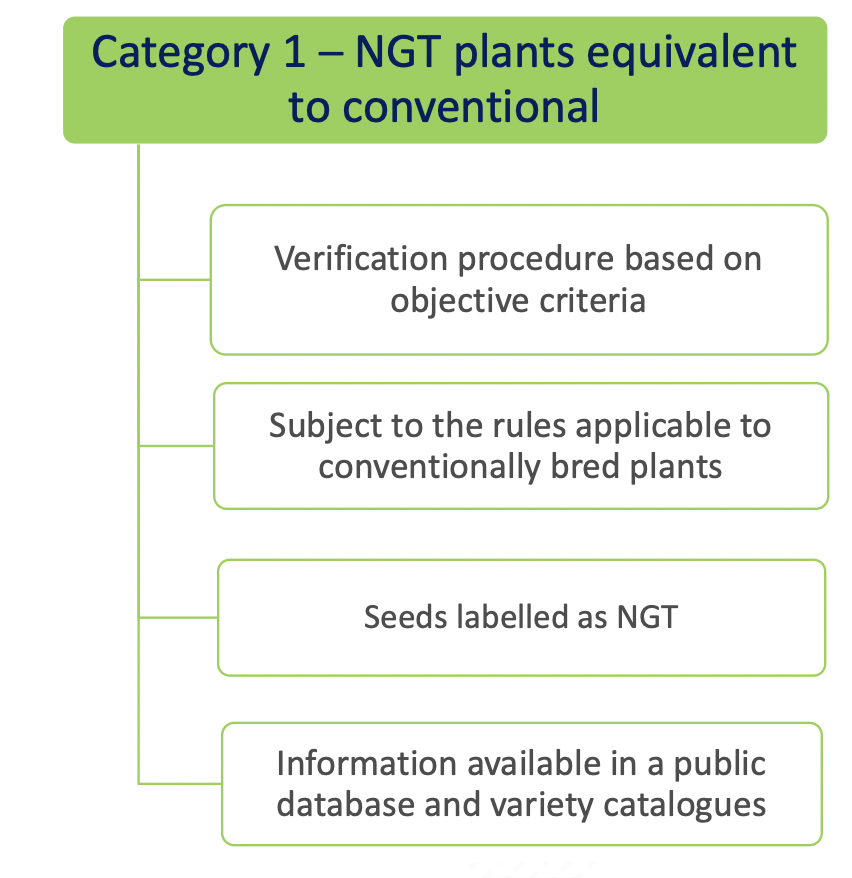
- Category 1 NGT plants: these plants are considered equivalent to conventionally bred plants based on the criteria in Annex I
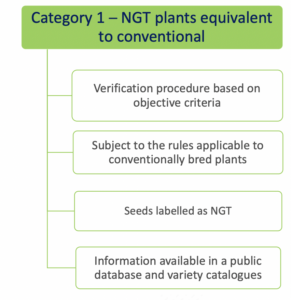 of the NGT Regulation and as such do not need to undergo complete GMO risk assessment. Instead, a notification to a national GMO authority or to EFSA is sufficient;
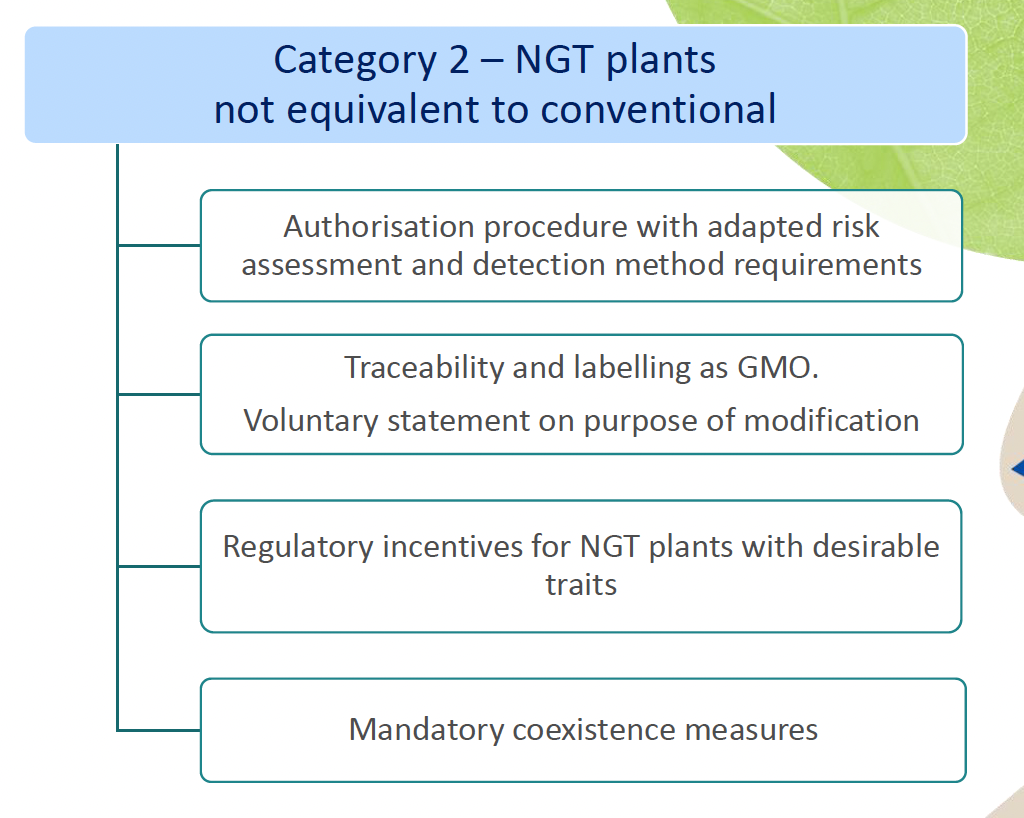
of the NGT Regulation and as such do not need to undergo complete GMO risk assessment. Instead, a notification to a national GMO authority or to EFSA is sufficient; - Category 2 NGT plants: these plants are considered as GMOs and as such are subject to the GMO rules for authorization, traceability and labeling, however according to a modified system of more targeted risk analysis.
 In addition to NGT plants, these regulations also apply to NGT products: that includes foods with ingredients made from such plants. This is why this proposed Regulation is so relevant for innovative food business operators.
In addition to NGT plants, these regulations also apply to NGT products: that includes foods with ingredients made from such plants. This is why this proposed Regulation is so relevant for innovative food business operators.
As mentioned above, for category 1 NGT plants, a notification procedure takes place and for category 2 NGT plants a full swing risk assessment. Both procedures involve a substantive assessment of whether plant material produced using targeted genetic techniques complies with the specifically developed rules for risk assessment. It is expected that the procedure for category 1 NGT products can be completed within a year, while the permit process for category 2 NGT foods is actually identical to that for regular GMOs. An innovation is that the NGT Regulation provides certain incentives for specific category 2 NGT foods that should speed up or simplify the assessment process.
Transparency regarding genetic engineering
Producers of organic products and advocacy organizations are concerned that, based on the NGT Regulation, genetically engineered food is walking into stores undetected. But is this concern justified? Based on the current text of this Regulation, both category 2 and even category 1 NGT products are excluded from organic production. Indeed, in the consultation process leading up to the NGT Regulation, the freedom of choice of consumers to buy food containing or not containing GMOs appeared to be an important issue. At the same time, the European Parliament considers the prohibition for organic farmers to use conventional-like category 1 NGT’s neither science-based, nor politically justifiable. It therefore calls in its draft report to create a fair level playing field and to only ban category 2 NGT plant material from organic production.
Category 1 NGT products listed in public database?
Whether or not allowed in organic production, there is no specific labeling requirement for Category 1 NGT products (Q 11 from EC Q&A). However, plant material with Category 1 NGT status must be included in a public database, expected to be similar to the Novel Food consultation database. In the Commisson’s proposal, plant reproduction material with Category 1 NGT status must additionally be labeled as such, with the identification number of the plant from which it is derived. At the level of production, this would provide an extra safeguard for distinguishing between conventionally grown crops and crops in which genetic engineering has been used. The European Parliament is however critical about this additional requirement in its draft report. It considers such discriminatory since category 1 NGT plants are conventional-like. According to the European Parliament, transparency and consumer choice are sufficiently ensured by disclosure in a public database. Therefore, even if the modifications proposed by the European Parliament will stand, there is no reason to believe that foods produced using CRISPR-Cas9 could go completely unnoticed.
Conclusion
With current changes in climate and constraints on available agricultural land with a growing world population, plant harvests will come under increasing pressure. NGTs are expected to meet the need to better equip plants for such challenges. The proposed NGT Regulation aims to simplify and thereby accelerate market access for such technology. For category 1 NGT products, except for a few open ends, this premise seems feasible in practice. A huge improvement with respect to current legislation is that the nature of the genetic change is decisive for risk evaluation, not the technique by which it is produced (product-based vs. process-based approach). For category 2 NGT products, market access is not expected to become much simpler based on the current proposal. Nevertheless, this proposal looks hopeful for plant innovations and thus for our food products of tomorrow. Also, there seems to be sufficient transparency to distinguish between crops produced with and without genetic engineering. My very bald hope would be that based on positive experiences with this regulation, the scope of this regulation will be extended to other fields, such as cellular agriculture and/or fermentation-based products. We will continue to follow closely how this legislation will develop.
Source images
- How CRISPR works: EU Parliament briefing on Plants produced by new genomic techniques
- Category 1 NGT plants: PPT DG Santé on Farm to Fork Strategy presented during EFFL Conference on 19 October 2023.
- Category 2 NGT plants: idem (2)
Thanks to my colleagues Jasmin Buijs and Max Baltussen for their valuable feed-back.
First ECJ ruling on current Novel Food Regulation
Posted: June 21, 2023 Filed under: Authors, Food, novel food Comments Off on First ECJ ruling on current Novel Food Regulation On May 25, the European Court of Justice (ECJ) ruled in a dispute arising between two manufacturers of food supplements. This is the first decision on the interpretation of the current Novel Food Regulation (applicable since Jan. 1, 2018). The dispute concerned the method of production of the functional ingredient spermidine. The dispute was hoped to clarify the question of what exactly a “new production process” under the Novel Food Regulation entails. The decision follows a request from a court in Austria, where this answer was needed to resolve a national dispute. This answer is also relevant to other countries, as the Novel Food Regulation applies EU-wide.
On May 25, the European Court of Justice (ECJ) ruled in a dispute arising between two manufacturers of food supplements. This is the first decision on the interpretation of the current Novel Food Regulation (applicable since Jan. 1, 2018). The dispute concerned the method of production of the functional ingredient spermidine. The dispute was hoped to clarify the question of what exactly a “new production process” under the Novel Food Regulation entails. The decision follows a request from a court in Austria, where this answer was needed to resolve a national dispute. This answer is also relevant to other countries, as the Novel Food Regulation applies EU-wide.
Why do people consume spermidine?
Spermidine supplementation takes place with a view to supporting cellular autophagy, or cell renewal. This could promote the prevention of cardiovascular disease, prevent food allergies, and control the symptoms of diabetes. It has even been suggested that spermidine could extend human lifespan by 5 to 7 years. So says Advocate General (A-G) Campos Sánchez-Bordona in his opinion on the case dated Jan. 19 this year, citing various scientific sources (see footnotes 12 and 13 of this opinion). The A-G is an important advisor to the ECJ, which generally follows his or her opinions.
Background to this dispute.
The dispute in Austria was brought by The Longevitiy Labs (TLL), which markets the spermidine supplement spermidineLIFE. TLL extracts spermidine from ungerminated wheat germs through a complex and expensive chemical process. TTL applied for and obtained an EU Novel Food authorization for this food product (see Union List, entry “spermidine-rich wheat germ extract“). Then competitor Optimize Health Solutions enters the market with its own spermidine supplement. This is produced using a much simpler and therefore cheaper production process based on hydroponic cultivation of buckwheat seeds in an aqueous solution with synthetic spermidine. After harvesting the seedlings are washed in water, dried and milled to obtain flour. TLL believes that Optimize Health also needs a Novel Food authorization to market its product. It instituted proceedings seeking an injunction against further marketing of Optimize Health’s product without such an authorization.
Optimize Health argues it does not need a Novel Food authorization, because its product is not covered by the Novel Food Regulation. It is a fully dried traditional food obtained without any selective novel extraction method. Furthermore, it states spermidine has been available in food supplements in the EU market for more than 25 years. The germination of the buckwheat seeds of which its product is made would be a primary production process, to which the EU Hygiene Regulation applies, not the Novel Food Regulation. Furthermore, the Novel Food Regulation does not apply because its product involves “plants prior to harvesting” and these do not count as food under the EU General Food Law Regulation. The Austrian court decided that clarification of European law was needed to resolve this dispute and referred five questions to the ECJ.
Questions from the Austrian referring court
National judges who refer questions to the ECJ usually go for as many anchors as possible and thinking three steps ahead. If a possible answer by the ECJ to one question leads to a follow-up question by the inquiring national court, that follow-up question will be submitted upfront as well. The downside of this system is that if answering the first question is the end of the matter, answering the remaining questions is no longer needed. That is what we call procedural economy. In summary, the referring Austrian court asks the following questions:
- Should a food consisting of flour from buckwheat seeds with a high spermidine content be qualified as Novel Food in the category “foods isolated from (parts of) plants?”
- If not, might it be a Novel Food because a new production process has been used and does that term include primary production processes?
- If it is a novel production process, does it matter whether that process was not applied at all or only not applied to spermidine?
- If primary production processes are not covered by the term “new production process”, is it correct that the process of germinating buckwheat seeds in a nutrient solution containing spermidine is not covered by the Novel Food Regulation because it does not apply to plants prior to harvesting?
- Does it make a difference whether the nutrient solution contains natural or synthetic spermidine?
Decision of the ECJ
The ECJ answers question 1 – be it with some reservation – in the affirmative. Optimize Health’s product is a Novel Food because there is no evidence that this product was used for human consumption to any significant degree within the EU before 1997. This short answer is somewhat disappointing, as it makes answering the remaining questions irrelevant. Still, the ECJ does share two interesting considerations regarding the current Novel Food Regulation. It points out that the concept of “history of safe use within the Union” is not defined with respect to Novel Foods that must undergo the full authorization process. However, it is defined with respect to traditional foods from third countries. These are products that have been used as food outside the EU for considerable time, such as chia seeds. These products are subject to the requirement that the safety of the food has been confirmed by compositional data and experience of continued use for at least 25 years in the usual diet of a significant number of people in at least one third country. The ECJ finds that this requirement must be applied by analogy to the spermidine in question and concludes that said data have not been provided.
Propagation methods vs. complete production process
Another consideration of the ECJ concerns the Novel Food category of “food isolated from (parts of) plants”. An exception applies to products made by non-traditional propagation methods, which do not result in significant changes in the composition or structure of the food in question. The ECJ ruled that a distinction must be made between:
(1) propagation processes of which the purpose is to produce new plants; and
(2) processes involving the entire production process of a food product.
The process applied to Optimize Health’s product to achieve a high spermidine content falls into the second category. In other words, a manufacturing technique to enrich a product is not the same as a propagation technique. The ECJ instructs the Austrian court to take this into account when deciding the case at the national level. This reduces the likelihood that the exception to the Novel Food category above will apply and puts the ECJ’s reservation into perspective. Good chance, therefore, that the Austrian national court will indeed determine that Optimize Health’s product is a Novel Food.
New production procedure according to the A-G
With the above answer, the question of the Austrian court has been answered and the ECJ does not get to the remaining four questions. It is of course unfortunate that we will not know the ECJ’s decision on this. Therefore, it is interesting to see how the A-G ruled on this. Well: according to the A-G, enriching buckwheat seeds with spermidine should be considered a new production process. He argues that bio-enrichment with spermidine changes the composition and nutritional value of the buckwheat seeds flour. Indeed, its spermidine content becomes 106 times higher than that of un-enriched buckwheat seeds. The A-G cites studies according to which a higher spermidine content may be beneficial to health, but which also indicate that too high an amount of spermidine could be harmful to cells.
The A-G therefore concludes that prior authorization of Optimize Health’s product is indispensable to ensure food safety and avoid risks to consumers. He also refers to products such as selenium-enriched mushrooms and mushrooms treated with ultraviolet light after harvest to increase their vitamin D2 content, where such authorization has also taken place. Furthermore, the A-G argues that the effects and thus the safety of a new production process should be assessed in each individual case and thus not in general. The same production process may affect one foodstuff differently from another.
Conclusion
This spermidine case clarifies the criterion that should be applied to determine whether a food has a history of safe use in the EU and thus qualifies or not as Novel Food. By the way, this is not entirely new – there has already been a guidance document from the European Commission “Human Consumption to a Significant Degree” since 1997 that argues essentially the same thing. However, when this judgment is considered in conjunction with the A-G’s opinion, it does provide relevant new information for determining how to establish whether there is a new production process.
This is the case if it is established that an applied process significantly alters the composition and nutritional value of a foodstuff compared to a foodstuff to which such a process has not been applied. Thus, in such a case, a food must obtain authorization under the Novel Food Regulation. The A-G recognizes that the question whether it is relevant if the production process has been previously applied to any foodstuff (rather than to the foodstuff specifically) cannot simply be answered based on the legal text. So that requires interpretation of the specific article of the Novel Food Regulation on new production processes in the light of its context and purpose. For now, we do not yet know whether the ECJ supports his interpretation. Hopefully we will find out in another case. It does sound plausible to me.
The FIC Regulation is due for renewal: How consumers can make healthy and sustainable food choices
Posted: April 21, 2023 Filed under: Authors, Food, Information Comments Off on The FIC Regulation is due for renewal: How consumers can make healthy and sustainable food choices The ‘farm to fork’ strategy calls for better communication to consumers about healthy and sustainable foods. To make this happen, EU legislation on food information to consumers is currently being revised.
The ‘farm to fork’ strategy calls for better communication to consumers about healthy and sustainable foods. To make this happen, EU legislation on food information to consumers is currently being revised.
The revision of the FIC Regulation covers front-of-pack nutrition labeling, establishment of nutrient profiles, origin labeling and date marking (‘best before’ / ‘use by’). The revision as prompted by the ‘farm-to-table’ strategy further relates to the labeling of alcoholic beverages, as announced in the European Cancer Control Plan.
Need for change
Our daily nutritional intake in Europe is not in line with national and international dietary recommendations. This leads to diet-related chronic diseases, such as diabetes and cardiovascular disease, with all kinds of consequences. According to the European Commission, this is partly because current labeling rules do not provide sufficient guidance for consumers to choose healthy foods.
Various proposals
As part of the legislative process, the European Commission published a so-called ‘Impact Assessment‘ at the end of 2020. The Impact Assessment sets out the labeling rules that need adjustment, and the different options for amendment as proposed by the European Commission. The Impact Assessment for tightened regulations on alcoholic beverages labeling was published later, in the summer of 2021. Below we discuss which changes to the FIC Regulation are proposed in the Impact Assessments and why, and touch upon the options presented for each topic. Later this year, the European Commission will follow up with a proposal for an amended regulation.
FOP labeling
Consumers do not always understand the nutritional information on packaging, which makes it difficult to choose healthy foods. To help consumers making better decisions, there are all kinds of voluntary initiatives for clear front-of-pack (FOP) nutrition labeling. Examples include the much discussed Nutri-Score, but also other initiatives such as the traffic light system (UK), the keyhole (Scandinavia) and the battery (Italy). However, these diverse FOP logos do not necessarily help European consumers as they do not provide equal access to information. The European Commission is concerned that this could lead to fragmentation of the single market, costs for food companies operating in several member states, and confusion among consumers. Harmonizing FOP logos and making them mandatory or not, are options on the table.
 Nutrient profiles
Nutrient profiles
Another proposed change concerns nutrition and health claims on unhealthy products. For example, consider the claim “source of fiber” for biscuits and “rich in vitamin C” for soft drinks. Such claims obscure the unhealthy profiles of these products, which leads to ‘health washing’ (term as introduced by the Dutch consumer association, see here). Nutrient profiles, i.e. thresholds for fats, sugars and/or salt above which the use of nutrition or health claims is restricted, could provide a solution to this. Such thresholds could help preventing consumer deception and create a level playing field for food companies. According to the Claims Regulation, nutrient profiles should already have been established as early as 2009, but this never materialized due to intense debate on the matter. The revision of the FIC Regulation revitalized this discussion. Based on stakeholder feedback on the Impact Assessment, the time seems ripe for change now.
Origin labeling
There is a growing demand from consumers to know the origin of food products. This allows consumers to make more sustainable choices, such as by choosing local products. Origin labeling is already mandatory for certain types of meat, among others. In the absence of harmonized rules regarding other food categories, a number of member states developed national rules for this purpose. Rules differentiating from member state to member state however lead to unequal access to information within the EU and fragments the internal market. The European Commission is therefore investigating whether and, if so, how, European rules on origin labeling could be further extended. This could mean an expansion of the product groups for which origin indication is required. Current discussions also include the production phase to which the indication should refer and the area size referred to (EU / non-EU, or e.g. country or regional level).
Date marking
We currently have two types of date marking. Whereas ‘use by’ refers to the expiry date for food safety reasons, ‘best before’ refers to the date by which the food retains its optimum quality. After this date, color variations may occur, for example, but the product is still safe to eat. The Impact Assessment states that less than one in two consumers understand the meaning of the two date markings. As a result, a lot of food ends up in the waste bin unnecessarily. By estimate 10% of the 88 million tons of food that is wasted annually is linked to date marking. In the context of improving sustainability by reducing food waste, the Impact Assessment presents options to educate consumers about date marking. It also suggests the possibility of removing the ‘best before’ quality date where this has little or no added value.
 Labeling of alcoholic beverages
Labeling of alcoholic beverages
Health damage from alcohol is a serious public health issue in the EU. To reduce alcohol consumption, it is important to inform consumers about what is actually in alcoholic beverages, both in terms of ingredients and nutrients. Currently, this is not mandatory for alcoholic beverages with an alcohol volume above 1.2%. Having said that, there are various initiatives for better information provision on alcohol, such as self-regulation within the beer and spirits sectors. To create a level playing field for operators, the European Commission communicated among others a proposal to remove the aforementioned labeling exception for all alcoholic beverages. Some of the then required information could be communicated off-label via a QR code.
Follow-up steps
Interested parties were able to share feedback with the European Commission after the publication of the two Impact Assessments. To further prepare the legislative proposal, a public consultation took place from 13 December 2021 to 7 March 2022. This process was designed to gather further views, experiences and suggestions on food labeling from stakeholders. As many as 3224 citizens, companies, interest groups, public authorities and other parties filled out the questionnaire made available for this purpose. A large proportion of respondents expressed their support for a harmonized FOP logo, as well as for improved terminology or a visual presentation of date marking. The European Commission’s legislative proposal is expected later this year.
This blogpost has also been published in Dutch at VMT.nl. The author thanks Marie-Claire Evers for her translation of this blogpost into English.
EU Harmonized food packaging legislation is speeding up
Posted: January 3, 2023 Filed under: Authors, Food Comments Off on EU Harmonized food packaging legislation is speeding up Last October, AXON contributed to the 16th European Food and Feed Law Conference by a session on circular economy, waste, packaging law, alternative materials, and the Single-Use Plastics (SUP) Directive. While we were back then still waiting for proposals by the European Commission on packaging (waste) and bioplastics, these long-awaited proposals have now been published. This blogpost discusses the main take-aways from these recent European proposals and provides deeplinks to the texts involved.
Last October, AXON contributed to the 16th European Food and Feed Law Conference by a session on circular economy, waste, packaging law, alternative materials, and the Single-Use Plastics (SUP) Directive. While we were back then still waiting for proposals by the European Commission on packaging (waste) and bioplastics, these long-awaited proposals have now been published. This blogpost discusses the main take-aways from these recent European proposals and provides deeplinks to the texts involved.
Packaging problem
Packaging plays without a doubt a very important role in the placing on the market of food. It protects and preserves food, and therefore contributes to increased shelf life and reduced amounts of food waste. It also offers a way to communicate food information to the consumer. At the same time, packaging, just like anything else we create, leaves an impact on the environment. As communicated in the Green Deal, it is the EU’s ambition to lower our amount of packaging waste as part of the green transition.
Bio-plastics
In a search to meet the EU’s goals for a circular economy and climate-neutrality by 2050, bio-plastics are emerging on the market as alternatives for conventional plastics. Bio-plastics can bring several advantages such as making packaging production less dependent on fossil fuels and reducing litter as they may dissolve over time. Their use is however not without challenges as discussed in an earlier blogpost. To improve the understanding around these materials and to clarify where bio-plastics can bring genuine environmental benefits, the European Commission recently published a Communication towards other EU bodies on a policy framework for bio-plastics. As is already clear from existing rules on environmental claims, the Commission stresses that generic claims on packaging such as ‘bio-plastic’ should be avoided to stay away from greenwashing and misleading consumers. It furthermore proposes to only label bio-plastic packaging and other products as ‘biobased’, ‘biodegradable’ or ‘compostable’ when it meets certain conditions. The main take-aways for businesses are as follows:
dissolve over time. Their use is however not without challenges as discussed in an earlier blogpost. To improve the understanding around these materials and to clarify where bio-plastics can bring genuine environmental benefits, the European Commission recently published a Communication towards other EU bodies on a policy framework for bio-plastics. As is already clear from existing rules on environmental claims, the Commission stresses that generic claims on packaging such as ‘bio-plastic’ should be avoided to stay away from greenwashing and misleading consumers. It furthermore proposes to only label bio-plastic packaging and other products as ‘biobased’, ‘biodegradable’ or ‘compostable’ when it meets certain conditions. The main take-aways for businesses are as follows:
Biobased:
- specify the exact and measurable share of biobased plastic content in the packaging; and
- ensure that the biomass is sustainably sourced – priority should be given to the use of organic waste or by-products rather than to primary biomass.
Biodegradable:
- specify that biodegradable packaging should not be littered;
- do not label products covered under the SUP Directive (the scope of which we discussed in more detail here and here) and other short-lived applications and/or litter-prone packaging as biodegradable; and
- specify how long the product needs to biodegrade, under which circumstances and in what environment.
Compostable:
- label only industrially compostable plastics that comply with relevant standards – the Commission will request the revision of the European Standard EN 13432:2000 for this purpose;
- use industrially compostable plastics only if the environmental benefits are higher than their alternatives and if they do not have a negative impact on the quality of the compost, taking into account consumer behavior; and
- specify the way in which the packaging should be disposed of using pictograms.
The Communication on bio-plastics refers, where relevant, to the Commission proposal for a Packaging and Packaging Waste Regulation (PPWR). The aforementioned proposal was also published at the end of last year and is discussed next.
Packaging and Packaging Waste Regulation
Before diving into the Commission’s proposal for a PPWR, it is useful to provide some background on the legal framework for packaging waste as guided by the Waste Framework Directive (WFD).
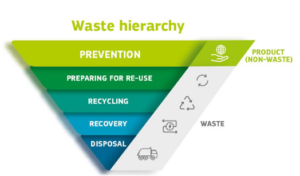 The WFD (currently under revision) introduces the waste hierarchy for waste management, which establishes an order of the preferred disposing route. Waste should in the first place be prevented/reduced. If this is not possible, re-use and thereafter recycling options should be looked into. Only in case this is (or is no longer) an option, energy recovery through incineration or ultimately landfill disposal should be considered. To reduce waste, the WFD also introduces the polluter pays principle and the extended producer responsibility, based on which the waste producer bears financial and/or organizational responsibility for the management of the waste stage at the end of a product’s life cycle.
The WFD (currently under revision) introduces the waste hierarchy for waste management, which establishes an order of the preferred disposing route. Waste should in the first place be prevented/reduced. If this is not possible, re-use and thereafter recycling options should be looked into. Only in case this is (or is no longer) an option, energy recovery through incineration or ultimately landfill disposal should be considered. To reduce waste, the WFD also introduces the polluter pays principle and the extended producer responsibility, based on which the waste producer bears financial and/or organizational responsibility for the management of the waste stage at the end of a product’s life cycle.
The proposal for the PPWR is the Commission’s answer to the revision of the current Packaging and Packaging Waste Directive (PPWD), which focuses on reducing, re-using and recycling packaging. The choice of legal instrument (a regulation rather than a directive) should facilitate a harmonized approach across the various EU Member States. The Regulation is however not an easy read. Although it contains only 65 legal articles, it includes many exceptions to the measures it proposes. For companies that want to get a feel of what to expect, we therefore compiled a list of the most important topics addressed in the Commission’s proposal for the PPWR.
- Requirements for packaging to be recyclable. From 2030, all packaging will have to be ‘designed for recycling’ in accordance with state-of-the-art collection, sorting and recycling processes. As of 2035, packaging must be ‘recycled at scale’, meaning that packaging must be sufficiently and effectively collected, sorted and recycled in practice. Further details on the design for recycling and recycling at scale requirements shall follow by delegated acts adopted by the Commission.
- Minimum amount of recycled plastic content. From 2030, plastic packaging shall contain certain minimum amounts (depending on the type of packaging) of recycled plastic content. These amounts shall further increase by 2040. Instructions as to the methodology for the calculation and verification of the percentage of recycled content will follow by an implementing act. Where information on the recycled content is communicated on the packaging, harmonized labels shall be used for such.
- Mandatory industrial composability for certain packaging. Think of coffee and tea bags or other units, sticky labels attached to fruit and vegetables, and very lightweight plastic carrier bags. The list of packaging that need to be industrially compostable may be extended in future. Packaging that could have been designed as re-usable shall not be presented as compostable.
- Sorting instructions. Labels with information on the material composition shall be applied on packaging to help consumers identifying the appropriate disposal route.
- Rules on re-use and refill. Certain economic operators in the take-away and beverage sector will be subject to targets on re-use and refill. Re-usable packaging must fulfill a set of criteria, including being part of a system for re-use. Information facilitating re-use must be provided on the packaging via a QR code or otherwise. In case of products offered through refill, end-consumers shall be provided with information to ensure safe and hygienic use of the product.
- Increased rules for manufacturers to demonstrate compliance. Manufacturers manufacturing packaging under their own name or trademark, or having packaging designed or manufactured for use with their products, are subject to increased rules to demonstrate compliance with the PPWR. Where a manufacturer is supplied with packaging or packaging materials from a third party, such supplier must provide the manufacturer with all information necessary to demonstrate conformity.
- New roles of economic operators. A system of checks & balances is introduced by giving authorized representatives, importers and distributors specific verification tasks to ensure packaging is placed on the market in accordance with the PPWR.
- Harmonized criteria for modulated extended producer responsibility fees. Financial contributions to be paid by producers (those making available packaging for the first time in the EU under their own name of trademark) to take responsibility for the management of packaging at their end-of-life shall be modulated based on the recyclability of the packaging and the presence of recycled plastic content.
- Reduced empty space in e-commerce and other packaging. The weight and volume of packaging shall be minimized as much as possible. The ratio of empty space in e-commerce and other pre-defined packaging in relation to the packaged product(s) shall not exceed 40%.
- Prohibition on packaging in certain formats and for certain purposes. This mainly concerns certain single-use applications such as in the HORECA sector and for small amounts of fresh fruit and vegetables. The list of prohibited packaging is presented in Annex V to the proposed PPWR and can be amended by delegated act.
- A deposit-return system for single-use plastic beverage bottles and beverage cans. Where these systems do not yet exist for packaging up to 3 liters, such shall be implemented by 2029. For other packaging, Member States are encouraged to voluntarily set up deposit-return or other systems to enable re-use or refill.
Take-away for businesses
As follows from the above, EU food packaging legislation is currently under revision: sustainable packaging will be the new norm. Although the Communication is not and the proposal for the PPWR is not yet binding law, companies involved with food packaging are advised to take the published information serious and to prepare for the enactment of official legislation. This does not only mean staying up-to-date with upcoming legal requirements applicable to the business at stake, but also being ready to involve and control partners in the supply chain through (revised) legal contracts.
Nomenclature for cultivated meat across Europe
Posted: October 31, 2022 Filed under: Authors, clean meat, cultivated meat, Food, novel food Comments Off on Nomenclature for cultivated meat across Europe This blogpost covers the recent GFI report European messaging for cultivated meat (GFI Report) as recently presented during the International Scientific Conference on Cultivated Meat (ISCCM).The further aim is providing the relevant regulatory context. Market authorisation is often mentioned as the delaying factor for market access of cellular agriculture-based products. If you are interested to know how the names and narratives by which these products are designated fit into the EU regulatory framework, read on!
This blogpost covers the recent GFI report European messaging for cultivated meat (GFI Report) as recently presented during the International Scientific Conference on Cultivated Meat (ISCCM).The further aim is providing the relevant regulatory context. Market authorisation is often mentioned as the delaying factor for market access of cellular agriculture-based products. If you are interested to know how the names and narratives by which these products are designated fit into the EU regulatory framework, read on!
Accelerated developments in cultivated meat field
Developments in the field of cellular agriculture have been tremendous since the first market approval of Eat Just’s hybrid cultivated chicken product in Singapore at the end of 2020. To name just a few:
- September 2021: Singapore Food Authority grants CMO Esco Aster a license to manufacture cultivated meat for commercial production. Meanwhile, several cultivated meat companies have concluded partnerships with Esco Aster for production purposes.
- October 2021: US Dept. of Agriculture announces an award of USD 10 million to Tufts University to set up a National Institute for Cellular Agriculture;
- October 2021: The Israeli Innovation Authority announces to invest an amount equalling USD 69 to establish four new public-private consortia, including one targeting cultivated meat;
- November 2021: opening of Upside Food’s 53.000 square foot production facility for cultivated meat;
- April 2022: The Dutch government agrees to invest through its National Growth Funds an amount of € 60 million to boost the formation of an ecosystem around cellular agriculture, representing the largest public funding in this field globally so far;
- October 2022: Mosa Meat announces construction of its 77 square cultivated meat campus.
- Upcoming event in November 2022: FAO Expert consultation in Singapore for gathering scientific advice on cell-based food products and food safety considerations.
For a comprehensive overview of the developments in the cultivated meat market, reference is made to GFI’s 2021 State of the Industry Report on Cultivated Meat and Seafood. This report also points out that investments in cultivated meat companies have grown from USD 420 million in 2020 to USD 1,8 billion in 2021. The sector clearly grows in interest and substance.
Market authorisation: where to start?
Quite a few of the cellular agriculture companies have their origin in the EU and in the UK. During the latest editions of KET Conference, the New Food Conference and the ISCCM, I witnessed however that most of them intend obtaining market approval in Singapore first, then in the United States of America and only afterwards in the European Union. Why is that? And it is justified based on the current EU regulatory framework?
EU: attractive but very diverse market
The European Union represents a market of more than 440 million consumers and thereby is a bigger market than for example the United States, counting currently over 330 million inhabitants. Based on this headcount alone it makes sense for each serious food business operator to consider the EU market for the launch of a new product. At the same time, the European Union consists of 27 Member States having their own cultural and culinary habits. This is exactly what is pointed out in the GFI Report. These varied backgrounds mostly require dedicated product communication. To a certain extent, some overlap in effective product communication in the four countries in which the research took place, was found as well.
Product communication vs. marketing
For clarity, there is a thin line between marketing and product communication, especially for pre-commercial companies that need to raise funds to get their product to the market. This was recognized by David Kay from Upside Foods in his ProVeg presentation, which you can watch here (starting at 26th minute). For the time being all cultivated meat companies, except perhaps East Just, are pre-commercial companies. Product communication means providing factual understandable information to the targeted public. Marketing means the direct or indirect recommendation of goods, services and/or concepts by on behalf of an advertiser, whether or not using third parties. The purpose of the GFI report is to develop positive, persuasive nomenclature and messaging for cultivated meat for each language and cultural context. In my view, the report thereby operates somewhere in the middle between product communication and marketing.
Rationale for common denominator
As recently acknowledged by the FAO, internationally harmonized terms to designate cultivated meat would be helpful to facilitate understanding worldwide:
“Cell-based food products are also referred to as “cultured” or “cultivated” followed by the name of the commodity, such as meat, chicken or fish while the process can also be called “cellular agriculture”. Given the various terminology in use for this technology, internationally harmonized terms for the food products and production processes would facilitate understanding at global level.”
Overlap in product communication
Back to the research performed by GFI. The very reason for the GFI Report is the current lack of consensus within the sector on the best nomenclatures and narratives to use. Negative framing of cultivated meat (which in some EU countries is already a reality) could prevent consumer acceptance of these products. GFI therefore tested which names and narratives worked well in each of France, Italy, Spain and Germany. Regarding the name, the GFI Report establishes that terms that loosely translate to “cultivated meat” are understood in all these countries and have a positive rather than a negative connotation. This comes down to “viande cultivée” / “carne coltivata” / “carne cultivada” / “kultiviertes Fleisch” or “Kulturfleisch”. As to the accompanying narratives, overall findings are that communication on cultivated meat should not be too technical. For example, reference to “cells” and “bioreactors” should generally be avoided, whereas analogies construed with existing food practices, such as the brewing of beer, work well.
Particularities for France, Italy, Spain and Germany
The research however also showed diverging results in the countries involved, both as regards the familiarity and the appreciation of cultivated meat. In France for instance, the terms “cells” and “bioreactor” are considered too reminiscent of a laboratory and too far removed from the language of food. In Italy “bioreactor” is even considered reminiscent of nuclear energy. In Spain, the terms “cells” and “bioreactors” are considered too scientific. In Germany on the other hand, the reference to “cells” is interpreted in an entirely different way. In this country, stating that cultivated meat stems from animal cells is interpreted that it tastes like conventional meat. In view of the market potential, in France 33% of the respondents indicate they would buy this food, whereas in Germany, Italy and Spain, these percentages are 57%, 55% and 65% respectively.
Do we have any examples from practice?
In an interview broadcasted on French television BFM Business on 5 October 2022, Nicolas Morin-Forest (NMF) delivers a fairly inspiring message on the cultivated foie gras of Gourmey. Whereas the TV station consistently refers to “viande synthèse”, NMF speaks of “viande culture” and stresses this is not a plant-based product (“Ce n’est pas du végétal”). Instead, he states, Gourmey delivers real animal protein with the same quality as animal protein in terms of taste (“C’est de la vraie protéine animale avec toutes les qualités gustatives des proteines animales”). He also mentions that with this product, it is no longer necessary to conclude any compromises; it associates culinary delight with the so-called protein transition (“Plus de compromis: le plaisir est au centre de l’assiette; au centre aussi de la transition alimentaire”). He finally points out that France can play a fundamental role in this protein transition, specifically based on its culinary foodprint and gastronomic history (“La France a un rôle fondamentale à jouer par notre patrimoine culinaire, par notre histoire gastronomique”).
Why are names and narratives of relevance for market authorisation?
The four researched countries are all important EU Member States, both in terms of head count and political influence. Germany is very influential at EU level and has the largest population of any EU country. Spain is reported to have strong influence over EU policy as well and has the highest meat intake in the EU. Both France and Italy have significant influence over EU agricultural policy. However, these latter two are the countries where we have seen the most hostile approach to non-conventional meat. In France for instance, there is ongoing litigation before the Conseil d’Etat concerning the prohibition of meaty names for non-conventional and alternative meat products.
Legal basis EU authorisation procedure
The position taken by all four countries will be of relevance during the authorisation procedure of cultivated meat in the EU. Cultivated meat – if produced without genetic modification – is regulated under the EU Novel Food Regulation. Contrary to legislation in Singapore (of very recent date) and in the US (still to be further shaped), this Regulation has already been in place since 1997 and was updated in 2018. The system is ready to receive applications when the companies are ready, too. Extensive EFSA guidance on the preparation of a Novel Food application is available.
Dynamics at the PAFF Committee
After EFSA makes available its safety evaluation regarding an application for authorisation of a cultivated meat product, the European Commission submits a draft implementing act to the PAFF Committee. This committee consists of representatives of each Member State and subsequently provides by qualified majority (i) a positive opinion, (ii) a negative opinion or (iii) no opinion at all. “Qualified majority” here means that 55% of the Member States vote in favour, representing at least 65 % of the EU population. The dynamics of this decision making procedure have been described in detail in the article Meat 3.0 – How Cultured Meat is Making its Way to the Market, which also provides for a flow chart at the end. In case of a negative opinion, one of the options is to escalate to the so-called Appeal Committee. In case of a negative opinion of the Appeal Committee, the EC shall not render an implementing act. In plain language: the application for authorisation of the cultivated meat product at stake will in such case be rejected.
Conclusion
Based on the above, It is quite easy to make the calculation that if for instance the representatives in the PAFF Committee from Germany (over 80 million inhabitants) or France (over 65 million inhabitants) do not vote in favour of an application for authorisation of a cultivated meat product, it will be difficult to reach the 65% threshold. Agreement on the name of product, understanding of the technology behind it, as well as the various benefits it could bring is therefore expected to be key for the evaluation procedure by the PAFF Committee. When these products make it to the EU market, they will be subject to the applicable legislation on food information and marketing. Until that time, it is of the essence that comprehensive product information reaches the relevant stake holders. It follows from the GFI Report that chefs and dieticians are best placed to deliver this message. In turn, the cultivated meat companies are in the position to provide them with relevant information. I can only encourage them to do so, if only to expedite market access in the EU.
————————-
Additional useful sources linked to this topic are ProVeg’s reports Communicating about cultured meat and The role of imagery in consumer perceptions of cultured meat (the latter targeting the UK specifically), each published in October 20222 and to be downloaded here.
————————
Foto credit: BioTech Foods
The era of nudging: EU’s proposals for mandatory front-of-pack labeling
Posted: June 13, 2022 Filed under: Advertising, Authors, Food, Information, Nutrition claims Comments Off on The era of nudging: EU’s proposals for mandatory front-of-pack labelingAs part of the EU Farm to Fork Strategy for a sustainable food system under the Green Deal, the European Commission agrees that nudging is necessary to guide the consumer to healthier and more sustainable food choices. This has translated into two impact assessments for mandatory front-of-pack (FOP) labeling. Various EU Member States and individual food business operators are however not waiting for harmonized EU FOP labeling and adopted the nutri-score (guiding health choices) and/or eco-score (guiding sustainability choices). This blogpost shows how these initiatives fit into the current and upcoming legal framework.
Nutri-score
 Nutri-score is a five-color nutrition label demonstrating the overall nutritional value of a food product front-of-pack. It allows consumers to compare various foods in a simple and fast way. The label is based on a scale of 5 color and letters, from a dark green “A” for the most healthy choice to a red “E” for the least healthy choice. Simply put, the algorithm behind nutri-score allocates positive points for favorable dietary components (fruits, vegetables, pulses nuts, fibers and proteins) and negative points for energy and unfavorable dietary components (saturated fatty acids, sugars and sodium). The total positive points are subsequently subtracted from the total negative points. The lower the score, the better the letter/color grade.
Nutri-score is a five-color nutrition label demonstrating the overall nutritional value of a food product front-of-pack. It allows consumers to compare various foods in a simple and fast way. The label is based on a scale of 5 color and letters, from a dark green “A” for the most healthy choice to a red “E” for the least healthy choice. Simply put, the algorithm behind nutri-score allocates positive points for favorable dietary components (fruits, vegetables, pulses nuts, fibers and proteins) and negative points for energy and unfavorable dietary components (saturated fatty acids, sugars and sodium). The total positive points are subsequently subtracted from the total negative points. The lower the score, the better the letter/color grade.
Fighting nutrition-related non-communicable diseases
The aim of nutri-score is to nudge the consumer into healthier food choices, and to stimulate the food industry to reformulate their recipes. This way, nutri-score should contribute substantially to a reduced burden of nutrition-related non-communicable diseases such as diabetes, cardiovascular diseases and some types of cancer.
Legal basis
From a legal perspective, nutri-score qualifies as voluntary food labeling in accordance with article 36 FIC Regulation. Food business operators that opt for using nutri-score are however obliged to use it for all foods they place on the market to avoid cherry picking. Moreover, a green “A” or “B” score additionally qualifies as a nutrition claim under the Claims Regulation. Since the claim is not listed in the annex to the Regulation, Member States adopting the nutri-score are subject to the notification procedure of article 23 of the Claims Regulation.
Algorithm changes
Countries that implemented nutri-score (France, Belgium, Luxembourg, Germany and Switzerland) or are willing to use it (Netherlands and Spain) join forces to ensure that nutri-score is in line with the national dietary guidelines. To coordinate such, the abovementioned countries established a Steering Committee and Scientific Committee in February 2021. The Steering Committee is composed of two representatives from national authorities in charge of the nutri-score implementation in each country; the Scientific Committee includes one or two independent experts nominated by each country involved. On March 7 last, the Scientific Committee published its interim report in which it proposes a methodology for modification of the nutri-score algorithm to handle problematic food categories (fats and oils, fish and seafood, whole grain products, salt, sugar, beverages, and dairy products). The Scientific Committee aims at providing a fully revised version of the nutri-score algorithm before the summer. The Steering Committee will have the final say in the recommendations proposed by the Scientific Committee and, where relevant, will elaborate a support document for food business operators to facilitate the appropriation of algorithm changes by the end of the year.
Developments at EU level
In the meantime, the European Commission held a public consultation to introduce standardized mandatory FOP nutrition labeling as part of the revision of the FIC Regulation within the EU’s Farm to Fork strategy. In its impact assessment, it listed five options:
- Baseline (“business as usual”) – it remains possible to voluntarily use a public or private, non-harmonized, FOP label.
- Nutrient- specific labels (numerical) – a harmonized FOP label such as the Italian Nutrinform Battery, providing numerical information on the content of macro nutrients and the energy value of a food, as well as the percentage of the daily refence intake that it makes up for.
- Nutrient-specific labels (color-coded) – a harmonized FOP label such as the UK Multiple Traffic Lights, which is similar to the numerical label but in addition uses colors to classify the content of nutrients as green, amber or red.
- Summary labels (endorsement logos) – a harmonized FOP label such as the Keyhole used in Sweden, which can be applied only to foods that comply with certain beneficial nutritional criteria.
- Summary labels (graded indicators) – a harmonized FOP label such as nutri-score, providing an appreciation of a product’s overall nutritional value through a graded indicator.
The harmonized FOP nutrition label as listed under 2 – 5 above could be either voluntary or mandatory, which is still subject to debate. The impact assessment also mentions the possibility of having a policy mix rather than using one preferred option. Next to the outcome of the public consultation, the European Commission will take into account the comprehensive review on FOP nutrition labeling schemes by the Joint Research Centre (2020) on EFSA’s recent scientific advise on nutrient profiling. A legislative proposal is expected in Q4 2022.
Eco-score
 Eco-score can be seen as the equivalent of nutri-score in the field of sustainability. Just like nutri-score, eco-score is a French initiative. It shows the consumer the environmental impact of a food, using the same presentation as nutri-score in terms of colors and letters. The food’s environmental impact is measured in two steps. First, the environmental footprint is calculated using the Product Environmental Footprint (PEF) method, which is based on a Life Cycle Assessment (LCA). The PEF method takes into account 16 different impact categories, such as ozone depletion, land use and climate change. This eventually translates into a score between 0 and 100. Thereafter, bonus points can be added up or minus points can be deduced from this score. These extra points (positive or negative) are based on 4 additional criteria: (1) food production methods as measured through attributed third-party sustainability credentials such as organic certification, fairtrade or MSC, (2) recyclability of the packaging, (3) the provenance of ingredients, and (4) the stay-away from biodiversity-related issues such as overfishing and deforestation. The LCA takes place at product category level; the allocated bonus and minus points are related to the individual product.
Eco-score can be seen as the equivalent of nutri-score in the field of sustainability. Just like nutri-score, eco-score is a French initiative. It shows the consumer the environmental impact of a food, using the same presentation as nutri-score in terms of colors and letters. The food’s environmental impact is measured in two steps. First, the environmental footprint is calculated using the Product Environmental Footprint (PEF) method, which is based on a Life Cycle Assessment (LCA). The PEF method takes into account 16 different impact categories, such as ozone depletion, land use and climate change. This eventually translates into a score between 0 and 100. Thereafter, bonus points can be added up or minus points can be deduced from this score. These extra points (positive or negative) are based on 4 additional criteria: (1) food production methods as measured through attributed third-party sustainability credentials such as organic certification, fairtrade or MSC, (2) recyclability of the packaging, (3) the provenance of ingredients, and (4) the stay-away from biodiversity-related issues such as overfishing and deforestation. The LCA takes place at product category level; the allocated bonus and minus points are related to the individual product.
Legal basis: currently no specific rules
There is not yet a legal framework specifically dedicated to environmental claims, let alone a legal definition thereof. Environmental claims are currently enforced based on general rules, guidelines and self-regulation within the legal framework of unfair commercial practices and misleading advertisements, as discussed in our earlier blogpost. Interestingly, these different sources produce different definitions of environmental claims. The definition thereof in the Dutch Code for Environmental Advertising is for example very broad and includes the eco-score as a claim related to the environmental factors connected with the product. It is however questionable whether an eco-score “C”, “D” or “E” would fall under the definition of a ‘green claim’ under the EC Guidance on the Unfair Commercial Practices Directive. This latter guidance namely refers to a positive environmental impact (which products with lower scores have not) or a lower damaging impact on the environment than competing products. Since the eco-score algorithm is largely based on product categories rather than individual products, it is not necessarily suitable for comparisons between competing products such as for example different fruit juices. Assuming that eco-score does qualify as an environmental claim, the following question is whether it is in conformity with the applicable rules. These rules are however not black and white and leave room for interpretation, especially since the number of enforcement cases is still rather low.
Future situation: EU harmonization of eco-score?
The ambiguity illustrated above may be over with in the near future, since the European Commission is working towards the harmonized use of a sustainability label under its Farm to Fork Strategy and the transition towards a sustainable food system. In its impact assessment, it lists the following options:
- Baseline (“business as usual”) – No specific new actions, though existing initiatives on environmental claims will be continued, such as the upcoming legislation on the substantiating of environmental footprint claims by use of the PEF or OEF (organizational environmental footprint) method.
- Voluntary approaches – No legislative initiatives but guidance and private initiatives such as codes of conducts.
- Reinforcing existing legislation – Development of sustainability labeling provisions related to more than one sustainability component (such as environmental and social sustainability) through existing sector-specific legislation (for example fisheries marketing standards).
- Voluntary EU sustainability label – Development of a voluntary harmonized sustainability label, either applicable to all foods or to foods that meet a certain sustainability standard only.
- Mandatory EU sustainability label – Development of a mandatory harmonized sustainability label, either for all foods placed on the EU market or mandatory for EU produced foods and voluntary for imported foods.
A legislative proposal is expected in Q4 2023. Until July 21 of this year, it is possible to contribute through the public consultation.
Meanwhile in the Member States….
 Member States seem not to be waiting for the legislative proposals of the European Commission. Instead, various Member States launched national initiatives on FOP sustainability labels and join forces to ensure that such labels will be implemented in a similar way throughout the EU. Taking the Netherlands as an example, the Ministry of Agriculture, Nature and Food Quality aims to implement a voluntary sustainability label for foods in the Netherlands by 2025. This goal forms part of the National Climate Agreement. Together with the food sector and other stakeholders, the Ministry is currently investigating how such a label for the Dutch market could look like. LCA’s based on the PEF-method are taken as a starting point for further development.
Member States seem not to be waiting for the legislative proposals of the European Commission. Instead, various Member States launched national initiatives on FOP sustainability labels and join forces to ensure that such labels will be implemented in a similar way throughout the EU. Taking the Netherlands as an example, the Ministry of Agriculture, Nature and Food Quality aims to implement a voluntary sustainability label for foods in the Netherlands by 2025. This goal forms part of the National Climate Agreement. Together with the food sector and other stakeholders, the Ministry is currently investigating how such a label for the Dutch market could look like. LCA’s based on the PEF-method are taken as a starting point for further development.
Conclusion
FOP labeling is a topic of conversation. Various initiatives on both national and European level are taking place simultaneously, in the hope that they will once come together as an EU harmonized label. We see different food businesses reacting to this situation differently. Where some opt for awaiting formal decisions at national level and instructions by the government, others are pioneering and experimenting with FOP labeling within the currently existing legal framework. Examples include the nutri-score pilot by Iglo and the full-fledged use of eco-score by Colruyt Group in Belgium. What about you? Are you a game changer or a laggerd?
Together with Lisa Gray from Iglo and Veerle Poppe from Colruyt Group, Jasmin Buijs presented this topic at the VMT Food Law Event on June 7 last.
This will change (soon): an update on environmental claims legal & policy framework
Posted: April 22, 2022 Filed under: Advertising, Authors, Food Comments Off on This will change (soon): an update on environmental claims legal & policy framework Many companies wonder what kind of environmental claim they can make for their product or packaging. Although there is no specific legal framework for this (yet), environmental claims fall under the general prohibition on misleading practices. Thanks to various developments at the European level, the legal framework for environmental claims is shaping up. Below is an update of the latest developments.
Many companies wonder what kind of environmental claim they can make for their product or packaging. Although there is no specific legal framework for this (yet), environmental claims fall under the general prohibition on misleading practices. Thanks to various developments at the European level, the legal framework for environmental claims is shaping up. Below is an update of the latest developments.
New guidance on the Unfair Commercial Practices Regulation
At the end of last year, the European Commission published an update of its guidance on the Unfair Commercial Practices Directive (“Revised Guidance”). The guidance provides amongst others an interpretation of the general prohibition on misleading practices in the aforementioned directive. Although the guidance has no legal status, it concerns an authoritative document. Judges and regulators such as the Netherlands Authority for Consumers and Markets (ACM) regularly refer to it. With regard to environmental claims, the Revised Guidance continues the line as set forth in its previous version from 2016. At certain points the text has been more detailed and more extensive examples are provided. However, the Revised Guidance also highlights a number of new points. The most striking additions are as follows.
Use of logos and labels
Environmental claims must be presented in a clear, specific, accurate and unambiguous manner. Simply placing a logo on a packaging will generally be insufficient to meet the above requirements since the average consumer cannot be expected to be familiar with the meaning thereof. This is not surprising considering that there are more than 100 sustainability logos in the EU. To prevent misleading practices, the Revised Guidance prescribes informing the consumer on the meaning of the logo, whether certification is done by a third party or not, and where further information can be found. These requirements are not absolutely new since they can also be found in the Guidelines sustainability claims issued by the ACM and the Code for Environmental Advertising by the Dutch self-regulatory organization of advertising (of which an update is expected this year). What is more is that the Revised Guidance sets forth additional requirements in case private instead of public quality marks are being used. The European Commission therefore seems to hint at a preference for public quality marks (e.g. EU Ecolabel, the Nordic Ecolabel ‘the Swan’ or the German ‘Blue Angel’) over private quality marks and to aim at a reduction of the total number of such marks. In this context, the ACM calls upon companies to use existing quality marks instead of developing their own.
Use asterisk for reference to additional information
It follows from the above that environmental claims (including logos) sometimes need further explanation to be well understood by the consumer. This is especially the case for general terms such as “sustainable” and “good for the environment”. A clarifying text should be placed as close as possible to the general claim, preferably right next to it. Another place may be chosen if there is no room for this, such as on the back of the packaging on which the claim is presented. An asterisk can be used to make the connection between the main claim and the additional information. A similar suggestion is made in the ACM Guidelines sustainability claims. The Revised Guidance states that when there is no room to specify the environmental claim, the claim should in principle be omitted.
Sharing information with competent authorities, but not with consumers
Environmental claims should be based on evidence that can be verified by the competent authorities. Authorities requesting such evidence are expected to take into account confidential information of the company making the environmental claim (such as certain input data for an LCA). The Revised Guidance emphasizes that the Unfair Commercial Practices Directive does not contain an obligation to provide such evidence to consumers (upon request). The ACM however takes a different position on this matter in its Guidelines sustainability claims, referring to the Guidelines for Making and Assessing Environmental Claims (2000) of the European Commission. The ACM advises placing evidence that substantiates the claim on a website and referring to it on (the packaging of) the product concerned. It also states that consumers should be provided with more information about the evidence concerned upon request. This raises the question how to deal with confidential information. A practical solution is to remove confidential information (provided this does not render the information concerned practically illegible), or to make a summary of the evidence for the claim available as a ‘consumer version’.
Completed public consultation on bio-plastics
Bio-plastics are a hot topic these days. These include ‘bio-based’, ‘biodegradable’ and ‘compostable’ plastics. Where bio-degradable plastics degrade and eventually dissolve due to changes in their chemical structure, the idea behind compostability is that a soil improver remains after the composting process. In contrast, bio-based plastics are plastics made from components derived from natural materials (as opposed to fossil-based materials). Bio-based plastics are not necessarily biodegradable or compostable. The EU is investigating the following policy areas with regard to bio-plastics.
Sustainability of natural materials vs. fossil resources
Plastics based on natural materials are often advertised as being more sustainable than fossil-based plastics. The question is, however, whether such natural materials offer real environmental benefits beyond a reduction in the use of fossil resources. To measure those benefits, the environmental impact of the full life cycle of such materials needs to be considered. This includes amongst other things the origin of the raw materials used. The use of arable land to grow natural materials for bio-plastics while this land could have been used for food is for example not necessarily ‘sustainable’.
Effective biodegradability and its role in a circular economy
 As far as biodegradability is concerned, it is being investigated whether and how plastics with this property fit into a circular economy. After all, there is little ‘circular’ about products that ‘dissolve’ in nature; reuse and recycling may offer better alternatives. It may at the same time be useful for certain product groups if they break down under specific conditions. An example of this could be agricultural film, which is not always removed completely after harvest and which is not easy to recycle after use due to contamination with soil. Similar questions arise with regard to compostable plastics. At best they break down into water and CO2, and therefore cannot contribute to soil improvement. Biodegradation of so-called compostable plastics could however also be useful for certain product groups. Think for example of compostable coffee and tea bags, which create a co-benefit when their use results into more coffee and tea being disposed in the green bin and subsequently composted.
As far as biodegradability is concerned, it is being investigated whether and how plastics with this property fit into a circular economy. After all, there is little ‘circular’ about products that ‘dissolve’ in nature; reuse and recycling may offer better alternatives. It may at the same time be useful for certain product groups if they break down under specific conditions. An example of this could be agricultural film, which is not always removed completely after harvest and which is not easy to recycle after use due to contamination with soil. Similar questions arise with regard to compostable plastics. At best they break down into water and CO2, and therefore cannot contribute to soil improvement. Biodegradation of so-called compostable plastics could however also be useful for certain product groups. Think for example of compostable coffee and tea bags, which create a co-benefit when their use results into more coffee and tea being disposed in the green bin and subsequently composted.
Misleading practices
 The prevention of misleading practices is also on the agenda. The European Commission recognizes that there is currently much confusion among consumers about bio-plastics. Stricter rules for the use of this and similar terms can prevent greenwashing. Moreover, it must be prevented that consumers interpret a biodegradability claim as a license to litter packaging with this characteristic.
The prevention of misleading practices is also on the agenda. The European Commission recognizes that there is currently much confusion among consumers about bio-plastics. Stricter rules for the use of this and similar terms can prevent greenwashing. Moreover, it must be prevented that consumers interpret a biodegradability claim as a license to litter packaging with this characteristic.
From 18 January to 15 March 2022, a public consultation took place at European level concerning the policy framework currently being under construction. Publication of the policy framework is planned for this summer.
European policy framework PEF and OEF
Environmental footprint claims, just like other environmental claims, need to be supported by evidence. There is however no harmonized method for substantiating such claims. Having said that, EU standard methods for the ecological footprint of products (PEF) or organizations (OEF) do exist since 2013. These methods focus on the measurement of the environmental performance of a product or organization over its entire life cycle using 16 environmental impact categories, including climate change, ozone depletion and water use. The European Commission is currently exploring the possibilities of putting more emphasis on the use of the abovementioned methods for the substantiation of environmental claims. The outcome of this study was planned for the first quarter of this year, but is still pending.
Ecodesign
There is even more happening in the field of sustainability in Brussels. On 30 March of this year, the European Commission adopted a proposal for a revised directive establishing rules for sustainable products (“Revised Ecodesign Directive”). Sustainable products will become the norm, with reusability, recyclability and energy efficiency being key concepts.
Food products are excluded from this European proposal on sustainable products. It is also unlikely that the Revised Ecodesign Directive will set specific rules for packaging materials meant for food contact. The rules to be established for packaging materials will namely be introduced via product-oriented requirements for products that do fall under the Revised Ecodesign Directive. This initiative nevertheless does mark a spot on the European sustainability horizon, which will also determine the direction for sustainability rules on packaging materials for foodstuffs.
Conclusion
Environmental claims are a topic of attention at national and European level. Food and food packaging businesses that make or wish to make environmental claims are therefore advised to keep an eye on current developments. Stay tuned!
Dairy under attack … and strikes back!
Posted: January 26, 2022 Filed under: Advertising, Authors, Food Comments Off on Dairy under attack … and strikes back! How to strike the right balance between freedom of speech and preventing unfair advertising targeting the dairy industry? This is the object of a Q4 2021 decision of the Appeal Board of the Dutch Advertising Code Committee, consisting of an advertising campaign on bus shelters in the Netherlands and a website campaign. If you are interested to know how broad the scope of “advertising” is and how a rather aggressive campaign was evaluated, continue reading. This post will be limited to the bus shelter campaign only since that already demonstrates the operation of the applicable legal framework.
How to strike the right balance between freedom of speech and preventing unfair advertising targeting the dairy industry? This is the object of a Q4 2021 decision of the Appeal Board of the Dutch Advertising Code Committee, consisting of an advertising campaign on bus shelters in the Netherlands and a website campaign. If you are interested to know how broad the scope of “advertising” is and how a rather aggressive campaign was evaluated, continue reading. This post will be limited to the bus shelter campaign only since that already demonstrates the operation of the applicable legal framework.
Bus shelter campaign attacking dairy industry
The bus shelter campaign showed several very explicit pictures demonstrating the misery life of young calves. Under the title “Do you need help quitting?” information was provided on the consequences of the dairy industry for these calves. The text on the various posters in the bus shelters stated the question “Do you need help quitting?”, followed by the following texts:
- Poster 1: “ “Dairy causes serious animal suffering. Calves are taken away from their mothers immediately after birth.”
- Poster 2: “Dairy is deadly. On a yearly basis, 1.5 million calves are slaughtered for dairy”.
- Poster 3: “Milk destroys more than you like. Calves are being fed artificial milk because the cow’s milk ended up in your cappuccino.”
The wording used above appeals to two well-known anti-smoking and anti-alcohol campaigns in the Netherlands, notably “quit smoking” and “alcohol destroys more than you like (your marriage for example).” The driver behind the anti-diary campaign is the Dutch organisation Animal & Law (“Dier & Recht”) that claims to be voice of the animals. This campaign was opposed by the Dutch dairy organization DairyNL (“ZuivelNL”), that aims to strengthen the Dutch dairy chain in a way that respects the environment and society. DairyNL considered this campaign misleading and therefore unfair.
Scope of the Dutch Advertising Code
The Netherlands knows a system of a self-regulation for advertising, embodied in the Dutch Advertising Code. Companies and other organisations can choose to submit to this system and the outcome (so-called recommendations) of cases based on complaints by whoever considers an advertisement misleading has a high degree of compliance (97 % in 2020). Animal & Law argued that their campaign was out of scope, as it did not envisage selling any products or services. Instead, it alleged to be a non-profit organisation that by public campaigns aims to improve the (legal) position of animals. Alternatively, it argued that if their campaign was captured by the scope of the Dutch Advertising Code, it would benefit from the principle of freedom of expression, as protected by the European Convention on Human Rights. Animal & Law overlooked however that the Dutch Advertising Code also captures ideas that are systematically recommended by an advertiser. The anti-dairy campaign by Animal & Law was found to meet this test. The Advertising Code Committee (“Reclame Code Commissie”) subsequently assessed for each bus shelter poster whether the information constituted misleading advertising.
Test for misleading advertising
The test applied is as follows: Advertising is unfair if it contravenes with standards of professional conduct and if it substantially disrupts or may disrupt the economic behaviour of the average consumer. One could consider this is exactly what Animal & Law was after and for a good cause. Under the Dutch Advertising Code however, aggressive advertising is at any rate considered unfair. Furthermore, all advertising containing incorrect or ambiguous information regarding specific aspects of the product at stake is considered misleading, if such information entices or may entice the average consumer to take a transaction decision (including refraining therefrom) that such consumer otherwise would not have made.
Poster 1
Just below the text “Do you need help quitting?” a milk carton is shown depicting a young calf being led away in a wheelbarrow followed by the text “Dairy causes serious animal suffering. Calves are taken away from their mothers immediately after birth.” All of this is depicted within a black framework, just like the health warning on a box of cigarettes. DairyNL argued, amongst other things, that according to the Netherlands Nutrition Centre, the consumption of dairy fits into a healthy diet, whereas the reference to quitting creates the impression dairy is very bad for human health. During the hearing, Animal & Law acknowledged that it is incorrect to state in general that dairy is not healthy. The Advertising Code Committee therefore considered the information re the quitting provided on poster 1 to be ambiguous and thus misleading. This results in unfair advertising.
Poster 2
This poster shows the same setting as poster 1, namely a milk carton depicting an earmarked calf surrounded by a black framework. According to DairyNL, in this context the text “Dairy is deadly. On a yearly basis, 1.5 million calves are slaughtered for dairy” can only be understood as a health warning. Furthermore, the absolute claim on the number of slaughtered calves creates the impression that this slaughter can only be attributed to the dairy industry, whereas it also serves meat production. Animal & Law had responded that calves are merely a by-product of dairy. It had used the warning that “dairy is deadly” to awaken the conscience of consumer. Obviously, the consumer understands the calf is going to die, not the consumer. The Advertising Code Committee considered this warning to be misleading, for the same reason as mentioned above. This part of the advertising was therefore considered unfair. The complaint regarding the slaughter was strikingly not addressed. I deduce therefrom that at any rate it was not concurred with.
Poster 3
On this poster, a calf is shown behind the bars of its small cage. DairyNL argued the setting of this poster (identical as described above) and the text “Milk destroys more than you like” links milk to two health hazard products, i.e. cigarettes and alcohol. DairyNL also argued that the claim “Calves are being fed artificial milk because the cow’s milk ended up in your cappuccino” creates the impression calves only drink artificial milk. Animal & Law refuted this complaint by explaining the claim does not state that calves only drink artificial milk. Animal & Law did not deny calves are being fed colostrum right after birth. However, this only takes 2 days, whereas the natural weaning period of calves amounts to 6 – 12 months. Here the Advertising Code Committee did not consider DairyNL’s claim founded, as DairyNL did not dispute that after a few days, the calves are being fed artificial milk instead of cow’s milk.
Conclusion
So what is the result of all this and what can we learn from this advertising decision? If the campaign of Animal & Law was mainly after shock and awe, it certainly succeeded. The pictures of the calves shown in the context of health warnings for cigarettes did not miss their effect. Potentially, a number of consumers became aware of certain facts it did not realise before. But will these consumers say a definite no to dairy? That’s the question, as the campaign may also have a counterproductive effect. Personally, I expect the chances of a seductive dairy alternative much higher for inducing consumers to eat no more (or just less) dairy. The learning from this decision is that even if the freedom of expression is a major public good, it also has its limits. No purpose justifies providing incorrect or ambiguous information. This learning applies equally to those outlining the pro’s of dairy alternatives, but certainly also to those emphasizing the con’s of dairy.