A drink made from monk fruit – novel food or not?
Posted: August 27, 2024 Filed under: Authors, Food, novel food Comments Off on A drink made from monk fruit – novel food or not?UK High Court quashes FSA decision on novelty of monk fruit decoctions
Introduction
In the EU, food business operators (FBOs) have the responsibility to ensure that the food they are marketing is not unsafe. Most of the food products do not require prior market authorization, novel foods being one of the exceptions to the rule. For novel foods market authorization is granted based on an extensive safety evaluation by EFSA. If an FBO is unsure if its product / ingredient falls within the scope of the EU Novel Food Regulation, it can submit a voluntary consultation to a Member State, where the FBO intends to first market its product. The Member State competent authority will review the data submitted to establish if a history of use before 1997 can be established. In the affirmative, the product will not be considered novel. Examples of products not considered novel include pea protein concentrate and the Chinese pepper Capsicum chinense.
 This blogpost covers the 19 March 2024 decision of the UK High Court on the novelty of a monk fruit decoction evaluated during a national consultation. This procedure was initiated by Guilin GFS Monk Fruit Corporation (Guilin GFS) against a joint decision of the UK Food Standards Agency (FSA) and the Food Standards Scotland (FSS). Guilin GFS is a world leader producer and manufacturer of monk fruit decoctions, having a 50 % global market share. Monk fruit is a small sub-tropical melon originating from China. Monk fruit decoctions can be applied to a wide range of foods and drinks as a sweetener and is popular for its low-calorie profile.
This blogpost covers the 19 March 2024 decision of the UK High Court on the novelty of a monk fruit decoction evaluated during a national consultation. This procedure was initiated by Guilin GFS Monk Fruit Corporation (Guilin GFS) against a joint decision of the UK Food Standards Agency (FSA) and the Food Standards Scotland (FSS). Guilin GFS is a world leader producer and manufacturer of monk fruit decoctions, having a 50 % global market share. Monk fruit is a small sub-tropical melon originating from China. Monk fruit decoctions can be applied to a wide range of foods and drinks as a sweetener and is popular for its low-calorie profile.
Figure 1 monk fruit image taken from the movie shown at www.monkfruitcorp.com
Applicable test and EC Guidance
After Brexit, the United Kingdom has maintained the EU Novel Food Regulation. Therefore, this decision is also of relevance for EU Member States. Under this applicable framework, the relevant test is whether monk fruit decoctions were used for human consumption to a significant degree in the EU or in the UK prior to 1997 (so-called history of consumption or HoC test). To adduce proof of a HoC, guidance can be taken from the Commission Guidance on Consumption to a Significant Degree dating back to 1997 (EC Guidance). In this context, it is important to note that the EC Guidance itself recognizes the difficulties of proof of significant use by the passage of time. It explicitly states that the examples of use are by no means exhaustive.
Evidence adduced
To meet the test on a HoC, Guilin GFS had adduced substantive evidence, including the following.
- Certificates of origin re. the export of processed foods monk fruits from mainland China to the UK during the period of 1998 – 2000;
- Evidence from a qualitative study from 2018 comprising face to face interviews with 71 participants in the UK and the EU demonstrating that processed foods containing monk fruits were sold in the EU / UK prior to 1997;
- Evidence from over 1.000 questionnaires as part of a quantitative population sample study from 2020 in the UK amongst people from Chinese descent re. their purchases of processed monk fruit;
- A survey in UK / EU supermarkets re. the types of monk fruit products sold before 1998;
- Signed declarations from restaurant owners, FBOs and the London Chinatown Chinese Association attesting the sale and / or consumption of monk fruit decoctions in the UK / EU prior to 1997.
FSA and FFS decision and rationale
On 8 September 2022, FSA and FFS rendered the decision that Guilin GFS did not meet the HoC test (the Decision). FSA and FFS considered the evidence too small in samples and none of this evidence hit the box of “Very Good Evidence”. This is the type of evidence referred to in Table 3 to the EC Guidance reproduced below. The table at stake contains examples of evidence that might be adduced to meet the HoC test, such as invoices detailing the sale of the food product at stake, including evidence of large quantities of the sale in the EU (“Very Good Evidence”), mere invoices detailing the sale of the food at stake (“Good Evidence”) and magazine articles (“Supporting Evidence”). The main reason why FSA and FFS considered the evidence not up to standards was because invoices demonstrating sales of monk fruit decoctions prior to1997 were missing. Also, the FSA considered that personal testimonies were inherently incapable of demonstrating a significant HoC without verification of an independent source.
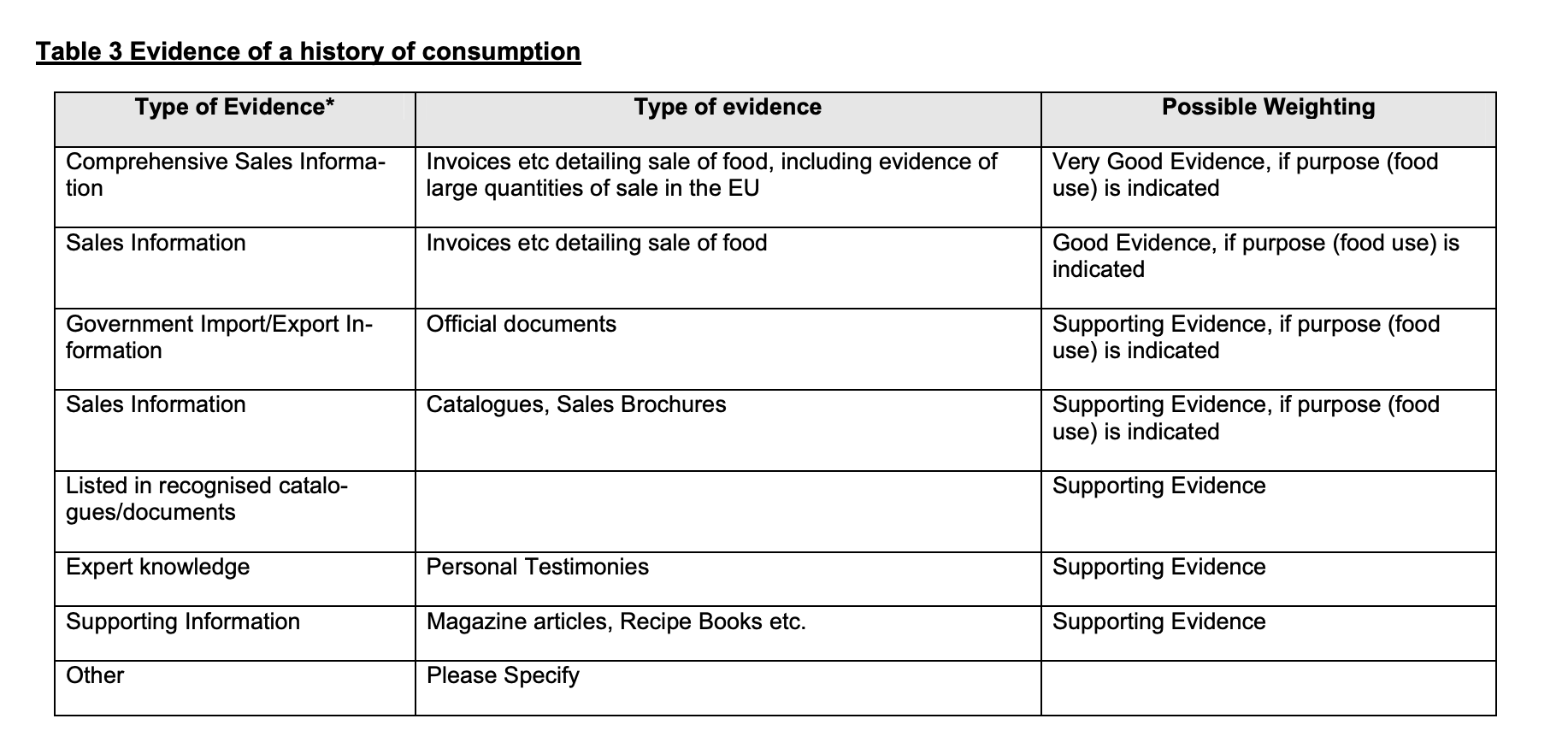
Figure 2: Table 3 to the EC Guidance on Consumption to a Significant Degree
Revision procedure and disclosure of FSA decision making process
Guilin GFS did not agree and initiated a revision procedure before the UK High Court on 2 December 2022, for which the hearing took place on 29 February 2024. The beauty of this procedure is that it provides full disclosure on all relevant documentation leading up to the Decision, including internal FSA correspondence conducted by its Novel Food policy advisors, as well as input from the FSA Social Sciences Team. This is an independent expert team, providing strategic advice to FSA. Contrary to the Novel Food FSA policy advisors, the Social Sciences Team considered the qualitative and the quantitative studies of Guilin to be reliable and robust. The Social Sciences Team did have a few verification requests to FSA’s Novel Food policy officers. However, according to Judge Calver, FSA’s Novel Food policy officers placed these questions out of statistic context to which they relate to validate the conclusion they reached earlier. Also, FSA’s Novel Food policy officers did not ask clarification questions to Guilin GFS when they reached their conclusion that monk fruit concoctions should be considered novel within the UK. Strikingly enough, they even reached this conclusion before the official validation of the dossier.
Debate during revision procedure
Guilin GFS opposed the Decision based on three legal grounds.
(1) It is incorrect that evidentiary requirements of the test for novelty under the Novel Food Regulation could not be met in the absence of pre-1997 sales invoices and data.
(2) It is incorrect that personal testimonies were categorically incapable to demonstrate a significant HoC unless verified by independent source.
(3) It is incorrect that it was necessary to demonstrate that monk fruit was consumed “exclusively for food uses”.
In reply, the Agencies served four witness statements from Novel Food policy advisors to “elucidate the reasons for the Decision.” Justice Calver states that based on the fact that these statements were made 15 months after the Decision, these need to be carefully scrutinized as there is a temptation to bolster and rationalize the Decision under challenge. Evidence that goes beyond elucidation and clarification is not permitted. When evaluating the witness statements, Justice Calver concludes the following.
Ad (1) The Agencies applied Table 3 to the EC Guidance too rigidly. Instead of considering “the whole picture” of evidence submitted by Guilin GFS, the Agencies saw that according to Table 3 the study results submitted by Guilin GFS did not qualify as “Very Good Evidence” or “Good Evidence”. They have then classified them as amounting “only” to “supporting evidence” “as defined in the Guidance”. This sentence makes clear that the Agencies consider anything other than invoices pre-1997 to be an inferior type of evidence as a category, amounting only to supporting evidence, which is not sufficient in itself to demonstrate a significant HoC.
Ad (2) The Agencies erroneously take the view that the evidence submitted by Guilin GFS’s should have been independently verified. There is no such requirement in the law or in the EC Guidance. Furthermore, the studies handed in by Guilin GFS had been qualified as reliable and robust by the Social Sciences Team. Also, with respect to personal testimonies, it is hard to conceive how these should be verified by third parties. As rightfully pointed out by Guilin GFS, such a requirement would frequently render personal testimonies redundant.
As a result, Judge Calver accepts grounds (1) and (2) and he considers the Decision flawed at these points. Moreover, he does not accept the four witness statements, as they try to re-write the reasoning in the Decision, which cannot stand with the wording of that document itself.
Ad (3) Finally, Judge Calver rules that the Agencies misapplied the relevant test when stating in the Decision that it was necessary to demonstrate that monk fruit was consumed “exclusively for food uses. The fact that the evidence submitted by Guilin GFS demonstrated a mixed use of monk fruit, in addition to food uses also including plant based medicinal products and even toothpaste, does not preclude establishing food use prior to 1997 in the EU or in the UK.
As a result, the claim for judicial review succeeds, the Decision is quashed, and the Agencies are ordered to re-consider the Claimant’s application in the light of this judgement. We can now see in the press that the Irish Food Safety Authority is reconsidering the novel food status of monk fruit extract and also the UK’s Food Standards Agency (FSA) has changed its stance regarding these products. The FSA now concludes that monk fruit concoctions are not a novel food, meaning that they can be used in food and beverages marketed in the UK. Reliable sources informed me that in the next weeks the Irish authorities – on behalf of the EU – are expected to issue a similar opinion to the UK.
Does this UK decision also apply for the EU?
In my opinion, this question should be answered positively. Rationale: according to the EC Guidance, an “established history of food use to a significant degree in at least one EU Member State is sufficient to exclude the food from the scope of Regulation (EU) 258/97.” The United Kingdom was an EU Member State during the period to which the evidence collected by Guilin GFS relates. The fact that Brexit occurred in 2020 does not change this. This also follows from the following statement in the EC Guidance: “The deadline 15 May 1997 is applicable to all Member States, irrespective from the date of accession to the EU.” By analogy, the same should apply in case of withdrawal of a Member State from the EU.
What can we learn from this decision?
In the first place, that it is a tough job to collect relevant evidence to establish a history of food use prior to 15 May 1997 in the EU (or the UK), especially since we are more and more moving away from this date. But it is doable. When looking at Guilin GFS, we can see that collecting product information, in combination with qualitative and quantitative data and affidavits can actually help to succeed in establishing such history of food use. Provided of course, you have a skilled lawyer at work to present your case. In the case at hand, the lawyers at work did an excellent job. One of these skilled lawyers is Brian Kelly, with whom I have been working together for more than 10 years now. Kudos to Brian!
First ECJ ruling on current Novel Food Regulation
Posted: June 21, 2023 Filed under: Authors, Food, novel food Comments Off on First ECJ ruling on current Novel Food Regulation On May 25, the European Court of Justice (ECJ) ruled in a dispute arising between two manufacturers of food supplements. This is the first decision on the interpretation of the current Novel Food Regulation (applicable since Jan. 1, 2018). The dispute concerned the method of production of the functional ingredient spermidine. The dispute was hoped to clarify the question of what exactly a “new production process” under the Novel Food Regulation entails. The decision follows a request from a court in Austria, where this answer was needed to resolve a national dispute. This answer is also relevant to other countries, as the Novel Food Regulation applies EU-wide.
On May 25, the European Court of Justice (ECJ) ruled in a dispute arising between two manufacturers of food supplements. This is the first decision on the interpretation of the current Novel Food Regulation (applicable since Jan. 1, 2018). The dispute concerned the method of production of the functional ingredient spermidine. The dispute was hoped to clarify the question of what exactly a “new production process” under the Novel Food Regulation entails. The decision follows a request from a court in Austria, where this answer was needed to resolve a national dispute. This answer is also relevant to other countries, as the Novel Food Regulation applies EU-wide.
Why do people consume spermidine?
Spermidine supplementation takes place with a view to supporting cellular autophagy, or cell renewal. This could promote the prevention of cardiovascular disease, prevent food allergies, and control the symptoms of diabetes. It has even been suggested that spermidine could extend human lifespan by 5 to 7 years. So says Advocate General (A-G) Campos Sánchez-Bordona in his opinion on the case dated Jan. 19 this year, citing various scientific sources (see footnotes 12 and 13 of this opinion). The A-G is an important advisor to the ECJ, which generally follows his or her opinions.
Background to this dispute.
The dispute in Austria was brought by The Longevitiy Labs (TLL), which markets the spermidine supplement spermidineLIFE. TLL extracts spermidine from ungerminated wheat germs through a complex and expensive chemical process. TTL applied for and obtained an EU Novel Food authorization for this food product (see Union List, entry “spermidine-rich wheat germ extract“). Then competitor Optimize Health Solutions enters the market with its own spermidine supplement. This is produced using a much simpler and therefore cheaper production process based on hydroponic cultivation of buckwheat seeds in an aqueous solution with synthetic spermidine. After harvesting the seedlings are washed in water, dried and milled to obtain flour. TLL believes that Optimize Health also needs a Novel Food authorization to market its product. It instituted proceedings seeking an injunction against further marketing of Optimize Health’s product without such an authorization.
Optimize Health argues it does not need a Novel Food authorization, because its product is not covered by the Novel Food Regulation. It is a fully dried traditional food obtained without any selective novel extraction method. Furthermore, it states spermidine has been available in food supplements in the EU market for more than 25 years. The germination of the buckwheat seeds of which its product is made would be a primary production process, to which the EU Hygiene Regulation applies, not the Novel Food Regulation. Furthermore, the Novel Food Regulation does not apply because its product involves “plants prior to harvesting” and these do not count as food under the EU General Food Law Regulation. The Austrian court decided that clarification of European law was needed to resolve this dispute and referred five questions to the ECJ.
Questions from the Austrian referring court
National judges who refer questions to the ECJ usually go for as many anchors as possible and thinking three steps ahead. If a possible answer by the ECJ to one question leads to a follow-up question by the inquiring national court, that follow-up question will be submitted upfront as well. The downside of this system is that if answering the first question is the end of the matter, answering the remaining questions is no longer needed. That is what we call procedural economy. In summary, the referring Austrian court asks the following questions:
- Should a food consisting of flour from buckwheat seeds with a high spermidine content be qualified as Novel Food in the category “foods isolated from (parts of) plants?”
- If not, might it be a Novel Food because a new production process has been used and does that term include primary production processes?
- If it is a novel production process, does it matter whether that process was not applied at all or only not applied to spermidine?
- If primary production processes are not covered by the term “new production process”, is it correct that the process of germinating buckwheat seeds in a nutrient solution containing spermidine is not covered by the Novel Food Regulation because it does not apply to plants prior to harvesting?
- Does it make a difference whether the nutrient solution contains natural or synthetic spermidine?
Decision of the ECJ
The ECJ answers question 1 – be it with some reservation – in the affirmative. Optimize Health’s product is a Novel Food because there is no evidence that this product was used for human consumption to any significant degree within the EU before 1997. This short answer is somewhat disappointing, as it makes answering the remaining questions irrelevant. Still, the ECJ does share two interesting considerations regarding the current Novel Food Regulation. It points out that the concept of “history of safe use within the Union” is not defined with respect to Novel Foods that must undergo the full authorization process. However, it is defined with respect to traditional foods from third countries. These are products that have been used as food outside the EU for considerable time, such as chia seeds. These products are subject to the requirement that the safety of the food has been confirmed by compositional data and experience of continued use for at least 25 years in the usual diet of a significant number of people in at least one third country. The ECJ finds that this requirement must be applied by analogy to the spermidine in question and concludes that said data have not been provided.
Propagation methods vs. complete production process
Another consideration of the ECJ concerns the Novel Food category of “food isolated from (parts of) plants”. An exception applies to products made by non-traditional propagation methods, which do not result in significant changes in the composition or structure of the food in question. The ECJ ruled that a distinction must be made between:
(1) propagation processes of which the purpose is to produce new plants; and
(2) processes involving the entire production process of a food product.
The process applied to Optimize Health’s product to achieve a high spermidine content falls into the second category. In other words, a manufacturing technique to enrich a product is not the same as a propagation technique. The ECJ instructs the Austrian court to take this into account when deciding the case at the national level. This reduces the likelihood that the exception to the Novel Food category above will apply and puts the ECJ’s reservation into perspective. Good chance, therefore, that the Austrian national court will indeed determine that Optimize Health’s product is a Novel Food.
New production procedure according to the A-G
With the above answer, the question of the Austrian court has been answered and the ECJ does not get to the remaining four questions. It is of course unfortunate that we will not know the ECJ’s decision on this. Therefore, it is interesting to see how the A-G ruled on this. Well: according to the A-G, enriching buckwheat seeds with spermidine should be considered a new production process. He argues that bio-enrichment with spermidine changes the composition and nutritional value of the buckwheat seeds flour. Indeed, its spermidine content becomes 106 times higher than that of un-enriched buckwheat seeds. The A-G cites studies according to which a higher spermidine content may be beneficial to health, but which also indicate that too high an amount of spermidine could be harmful to cells.
The A-G therefore concludes that prior authorization of Optimize Health’s product is indispensable to ensure food safety and avoid risks to consumers. He also refers to products such as selenium-enriched mushrooms and mushrooms treated with ultraviolet light after harvest to increase their vitamin D2 content, where such authorization has also taken place. Furthermore, the A-G argues that the effects and thus the safety of a new production process should be assessed in each individual case and thus not in general. The same production process may affect one foodstuff differently from another.
Conclusion
This spermidine case clarifies the criterion that should be applied to determine whether a food has a history of safe use in the EU and thus qualifies or not as Novel Food. By the way, this is not entirely new – there has already been a guidance document from the European Commission “Human Consumption to a Significant Degree” since 1997 that argues essentially the same thing. However, when this judgment is considered in conjunction with the A-G’s opinion, it does provide relevant new information for determining how to establish whether there is a new production process.
This is the case if it is established that an applied process significantly alters the composition and nutritional value of a foodstuff compared to a foodstuff to which such a process has not been applied. Thus, in such a case, a food must obtain authorization under the Novel Food Regulation. The A-G recognizes that the question whether it is relevant if the production process has been previously applied to any foodstuff (rather than to the foodstuff specifically) cannot simply be answered based on the legal text. So that requires interpretation of the specific article of the Novel Food Regulation on new production processes in the light of its context and purpose. For now, we do not yet know whether the ECJ supports his interpretation. Hopefully we will find out in another case. It does sound plausible to me.
Nomenclature for cultivated meat across Europe
Posted: October 31, 2022 Filed under: Authors, clean meat, cultivated meat, Food, novel food Comments Off on Nomenclature for cultivated meat across Europe This blogpost covers the recent GFI report European messaging for cultivated meat (GFI Report) as recently presented during the International Scientific Conference on Cultivated Meat (ISCCM).The further aim is providing the relevant regulatory context. Market authorisation is often mentioned as the delaying factor for market access of cellular agriculture-based products. If you are interested to know how the names and narratives by which these products are designated fit into the EU regulatory framework, read on!
This blogpost covers the recent GFI report European messaging for cultivated meat (GFI Report) as recently presented during the International Scientific Conference on Cultivated Meat (ISCCM).The further aim is providing the relevant regulatory context. Market authorisation is often mentioned as the delaying factor for market access of cellular agriculture-based products. If you are interested to know how the names and narratives by which these products are designated fit into the EU regulatory framework, read on!
Accelerated developments in cultivated meat field
Developments in the field of cellular agriculture have been tremendous since the first market approval of Eat Just’s hybrid cultivated chicken product in Singapore at the end of 2020. To name just a few:
- September 2021: Singapore Food Authority grants CMO Esco Aster a license to manufacture cultivated meat for commercial production. Meanwhile, several cultivated meat companies have concluded partnerships with Esco Aster for production purposes.
- October 2021: US Dept. of Agriculture announces an award of USD 10 million to Tufts University to set up a National Institute for Cellular Agriculture;
- October 2021: The Israeli Innovation Authority announces to invest an amount equalling USD 69 to establish four new public-private consortia, including one targeting cultivated meat;
- November 2021: opening of Upside Food’s 53.000 square foot production facility for cultivated meat;
- April 2022: The Dutch government agrees to invest through its National Growth Funds an amount of € 60 million to boost the formation of an ecosystem around cellular agriculture, representing the largest public funding in this field globally so far;
- October 2022: Mosa Meat announces construction of its 77 square cultivated meat campus.
- Upcoming event in November 2022: FAO Expert consultation in Singapore for gathering scientific advice on cell-based food products and food safety considerations.
For a comprehensive overview of the developments in the cultivated meat market, reference is made to GFI’s 2021 State of the Industry Report on Cultivated Meat and Seafood. This report also points out that investments in cultivated meat companies have grown from USD 420 million in 2020 to USD 1,8 billion in 2021. The sector clearly grows in interest and substance.
Market authorisation: where to start?
Quite a few of the cellular agriculture companies have their origin in the EU and in the UK. During the latest editions of KET Conference, the New Food Conference and the ISCCM, I witnessed however that most of them intend obtaining market approval in Singapore first, then in the United States of America and only afterwards in the European Union. Why is that? And it is justified based on the current EU regulatory framework?
EU: attractive but very diverse market
The European Union represents a market of more than 440 million consumers and thereby is a bigger market than for example the United States, counting currently over 330 million inhabitants. Based on this headcount alone it makes sense for each serious food business operator to consider the EU market for the launch of a new product. At the same time, the European Union consists of 27 Member States having their own cultural and culinary habits. This is exactly what is pointed out in the GFI Report. These varied backgrounds mostly require dedicated product communication. To a certain extent, some overlap in effective product communication in the four countries in which the research took place, was found as well.
Product communication vs. marketing
For clarity, there is a thin line between marketing and product communication, especially for pre-commercial companies that need to raise funds to get their product to the market. This was recognized by David Kay from Upside Foods in his ProVeg presentation, which you can watch here (starting at 26th minute). For the time being all cultivated meat companies, except perhaps East Just, are pre-commercial companies. Product communication means providing factual understandable information to the targeted public. Marketing means the direct or indirect recommendation of goods, services and/or concepts by on behalf of an advertiser, whether or not using third parties. The purpose of the GFI report is to develop positive, persuasive nomenclature and messaging for cultivated meat for each language and cultural context. In my view, the report thereby operates somewhere in the middle between product communication and marketing.
Rationale for common denominator
As recently acknowledged by the FAO, internationally harmonized terms to designate cultivated meat would be helpful to facilitate understanding worldwide:
“Cell-based food products are also referred to as “cultured” or “cultivated” followed by the name of the commodity, such as meat, chicken or fish while the process can also be called “cellular agriculture”. Given the various terminology in use for this technology, internationally harmonized terms for the food products and production processes would facilitate understanding at global level.”
Overlap in product communication
Back to the research performed by GFI. The very reason for the GFI Report is the current lack of consensus within the sector on the best nomenclatures and narratives to use. Negative framing of cultivated meat (which in some EU countries is already a reality) could prevent consumer acceptance of these products. GFI therefore tested which names and narratives worked well in each of France, Italy, Spain and Germany. Regarding the name, the GFI Report establishes that terms that loosely translate to “cultivated meat” are understood in all these countries and have a positive rather than a negative connotation. This comes down to “viande cultivée” / “carne coltivata” / “carne cultivada” / “kultiviertes Fleisch” or “Kulturfleisch”. As to the accompanying narratives, overall findings are that communication on cultivated meat should not be too technical. For example, reference to “cells” and “bioreactors” should generally be avoided, whereas analogies construed with existing food practices, such as the brewing of beer, work well.
Particularities for France, Italy, Spain and Germany
The research however also showed diverging results in the countries involved, both as regards the familiarity and the appreciation of cultivated meat. In France for instance, the terms “cells” and “bioreactor” are considered too reminiscent of a laboratory and too far removed from the language of food. In Italy “bioreactor” is even considered reminiscent of nuclear energy. In Spain, the terms “cells” and “bioreactors” are considered too scientific. In Germany on the other hand, the reference to “cells” is interpreted in an entirely different way. In this country, stating that cultivated meat stems from animal cells is interpreted that it tastes like conventional meat. In view of the market potential, in France 33% of the respondents indicate they would buy this food, whereas in Germany, Italy and Spain, these percentages are 57%, 55% and 65% respectively.
Do we have any examples from practice?
In an interview broadcasted on French television BFM Business on 5 October 2022, Nicolas Morin-Forest (NMF) delivers a fairly inspiring message on the cultivated foie gras of Gourmey. Whereas the TV station consistently refers to “viande synthèse”, NMF speaks of “viande culture” and stresses this is not a plant-based product (“Ce n’est pas du végétal”). Instead, he states, Gourmey delivers real animal protein with the same quality as animal protein in terms of taste (“C’est de la vraie protéine animale avec toutes les qualités gustatives des proteines animales”). He also mentions that with this product, it is no longer necessary to conclude any compromises; it associates culinary delight with the so-called protein transition (“Plus de compromis: le plaisir est au centre de l’assiette; au centre aussi de la transition alimentaire”). He finally points out that France can play a fundamental role in this protein transition, specifically based on its culinary foodprint and gastronomic history (“La France a un rôle fondamentale à jouer par notre patrimoine culinaire, par notre histoire gastronomique”).
Why are names and narratives of relevance for market authorisation?
The four researched countries are all important EU Member States, both in terms of head count and political influence. Germany is very influential at EU level and has the largest population of any EU country. Spain is reported to have strong influence over EU policy as well and has the highest meat intake in the EU. Both France and Italy have significant influence over EU agricultural policy. However, these latter two are the countries where we have seen the most hostile approach to non-conventional meat. In France for instance, there is ongoing litigation before the Conseil d’Etat concerning the prohibition of meaty names for non-conventional and alternative meat products.
Legal basis EU authorisation procedure
The position taken by all four countries will be of relevance during the authorisation procedure of cultivated meat in the EU. Cultivated meat – if produced without genetic modification – is regulated under the EU Novel Food Regulation. Contrary to legislation in Singapore (of very recent date) and in the US (still to be further shaped), this Regulation has already been in place since 1997 and was updated in 2018. The system is ready to receive applications when the companies are ready, too. Extensive EFSA guidance on the preparation of a Novel Food application is available.
Dynamics at the PAFF Committee
After EFSA makes available its safety evaluation regarding an application for authorisation of a cultivated meat product, the European Commission submits a draft implementing act to the PAFF Committee. This committee consists of representatives of each Member State and subsequently provides by qualified majority (i) a positive opinion, (ii) a negative opinion or (iii) no opinion at all. “Qualified majority” here means that 55% of the Member States vote in favour, representing at least 65 % of the EU population. The dynamics of this decision making procedure have been described in detail in the article Meat 3.0 – How Cultured Meat is Making its Way to the Market, which also provides for a flow chart at the end. In case of a negative opinion, one of the options is to escalate to the so-called Appeal Committee. In case of a negative opinion of the Appeal Committee, the EC shall not render an implementing act. In plain language: the application for authorisation of the cultivated meat product at stake will in such case be rejected.
Conclusion
Based on the above, It is quite easy to make the calculation that if for instance the representatives in the PAFF Committee from Germany (over 80 million inhabitants) or France (over 65 million inhabitants) do not vote in favour of an application for authorisation of a cultivated meat product, it will be difficult to reach the 65% threshold. Agreement on the name of product, understanding of the technology behind it, as well as the various benefits it could bring is therefore expected to be key for the evaluation procedure by the PAFF Committee. When these products make it to the EU market, they will be subject to the applicable legislation on food information and marketing. Until that time, it is of the essence that comprehensive product information reaches the relevant stake holders. It follows from the GFI Report that chefs and dieticians are best placed to deliver this message. In turn, the cultivated meat companies are in the position to provide them with relevant information. I can only encourage them to do so, if only to expedite market access in the EU.
————————-
Additional useful sources linked to this topic are ProVeg’s reports Communicating about cultured meat and The role of imagery in consumer perceptions of cultured meat (the latter targeting the UK specifically), each published in October 20222 and to be downloaded here.
————————
Foto credit: BioTech Foods
Singapore cultured meat regulatory approval process compared to EU
Posted: April 26, 2021 Filed under: alternative protein, Authors, clean meat, Food, novel food Comments Off on Singapore cultured meat regulatory approval process compared to EU Cultured meat is making its way to the market globally. Recently, we saw this movie from Super Meat, serving cultured meat in a restaurant where in the same time cultured meat was actually being cultured. Earlier, tasting sessions by Aleph Farms of its cell-based steak were shared via social media. And at the end of last year, it was reported that Eat Just had gained regulatory approval to market its cultured chicken in Singapore. This made me (and I guess many others!) wonder how the regulatory approval process is set up in the Lion City. Last week, I had an interview with the Singapore Food Agency (SFA), the outcome of which I report in this blogpost. I also compare the information received to the regulatory system for authorisation of cultured meat in place in the EU.
Cultured meat is making its way to the market globally. Recently, we saw this movie from Super Meat, serving cultured meat in a restaurant where in the same time cultured meat was actually being cultured. Earlier, tasting sessions by Aleph Farms of its cell-based steak were shared via social media. And at the end of last year, it was reported that Eat Just had gained regulatory approval to market its cultured chicken in Singapore. This made me (and I guess many others!) wonder how the regulatory approval process is set up in the Lion City. Last week, I had an interview with the Singapore Food Agency (SFA), the outcome of which I report in this blogpost. I also compare the information received to the regulatory system for authorisation of cultured meat in place in the EU.
Which foods are considered Novel Foods in Singapore?
Before putting in place its Novel Foods regulatory framework in 2019, the SFA took inspiration from similar legislation applicable in other parts of the world, amongst others in the EU. This clearly transpires from the definition of a Novel Food. The SFA considers Novel Foods to be foods that do not have a history of safe use. This is the case if substances have not been consumed as an ongoing part of the (human) diet by a significant human population during at least 20 years. This definition is pretty similar to the one used in the EU, except that the EU definition uses (i) 15 May 1997 as a fixed point in time cut-off date and (ii) a closed-loop system of 10 Novel Food categories. In Singapore, just like in other places of the world, it goes without saying that cultured meat (“meat developed from animal cell culture”) requires prior market authorisation. Whereas in the EU this could also take place (depending on the production process applied) on the basis of a GMO authorisation, my understanding is that in Singapore the designated route is a Novel Food authorisation. In the EU, a distinction is made between foods containing GMOs and foods produced from GMOs on the one hand and food produced with GMOs on the other. Whereas the first two categories require GMO clearance, the third one does not.
Organisation Singapore Food Agency
Contrary to the EU, where EFSA operates as an external advisory body to the European Commission the National Centre for Food Science (NCFS) forms an integral part of the SFA. The NCFS consists of food inspection services and laboratory testing services and a representative thereof also took part in the interview. This setup and Singapore’s 5.7 million inhabitants (compared to over 445 million in the EU) potentially explains the shorter timelines during the regulatory approval process, as outlined below.
Regulatory Approval Process
The SFA acknowledges the science for producing cultured meat is still in an early stage. So far, it has set up a modest framework defining the Requirements for the Safety Assessment of Novel Foods. It however explicitly states the information required may change based on the developments on the science of producing cultured meat. The data required for evaluating the safety of Novel Foods largely correspond to the data required under the EU regulatory framework. They include information on the identity and the manufacturing process applied, as well as on the intended use and proposed levels of use. Toxicity studies (both in-vitro and in-vivo) and metabolism or toxicokinetic studies are requested “where relevant”. For cultured meat specifically, they include information on the cell lines, culture media and scaffolding materials used. Finally, any safety assessment reports conducted by the food safety authorities of Australia, Canada, New Zealand, Japan, the EU and the USA are considered relevant. For the EU, the SFA Guidance states that safety assessments conducted in accordance with the ESFA Guidance for submission of food additive evaluations would be accepted for review. This guidance is referred to in the EFSA Guidance for novel food applications in the context of toxicity testing and propagates a tiered approach for the type of safety parameters to be satisfied. This means that if no risk indicators are revealed in the studies performed in the first tier, no studies from the second or third tier need to be carried out.
Procedural aspects
The estimated timeline to complete an evaluation of a Novel Food in Singapore is said to be 3 – 6 months. During the interview however, SFA staff indicated this to be “slightly optimistic”. For the time being, the SFA has not yet put down any requirements regarding procedural aspects of a Novel Food application; this is still in the works. What struck me most during the interview is the informal and cooperative approach of the SFA. They seem to offer their assistance as a service rather than that they position themselves as rigid safety assessors. Also, they much encourage companies to consult SFA early in their product development process (referred to as “early-stage engagement”). This will enable mutual understanding of the production process of the cultured meat product at stake, as well as the applicable safety requirements. This may subsequently enable companies to make deliberate choices in their manufacturing setup. All of this is quite different in the EU, where interactions with EFSA is highly regulated and where detailed procedural regulations for submissions are in place, amongst others in view of protection of transparency in the safety evaluation process. Also, based on the Union studies database, it will be public what studies form the backbone of the safety evaluation for the Novel Food at stake.
Confidentiality vs. transparency
The notion of transparency was not mentioned once during my interview with the SFA. Instead, it was said that all information submitted during the application process is confidential. This may be advantageous for companies who want to make it quickly to the market. In the long run, and especially if authorisation of Novel Foods by the SFA will operate as a steppingstone for access of other Asian markets, I expect the need for a similar system as in place in the EU will arise.
Denomination
Can cultured meat actually be called meat in Singapore? This may seem to be a no-brainer, but this is a highly debated question in the EU, where also the use of typical dairy names for plant-based alternatives is prohibited. The designation “meat” for cultured meat products in the EU may be limited based on the Hygiene Regulation, that reserves the name of “meat” for “edible parts” of specific animals, such as beef and pork. Now that cultivated cells (or even stem-cells) may not necessarily qualify as such edible parts, this issue still needs to be resolved. When marketing cultured meat products, the SFA does not oppose the designation of “meat”, provided that it is accompanied by a qualifier, for example cultured meat. “Clean” meat will however be a no-go, as it might have negative connotations for traditional meat. The proposed solution here seems appropriate to me to adequately inform the consumer what type of product is offered for sale and sufficient to prevent any misleading. Since that is also the cornerstone of EU food information law, I would very much advocate a similar solution to apply in the EU.
Conclusion
Authorisation of a Novel Food such as cultured meat takes place in a relatively informal and accelerated, yet science-based way in Singapore. It is therefore not surprising that the world’s first cultured meat product obtained regulatory clearance there. For now, we have not seen any applications being submitted in any other territory, not in the US, not in the EU and not even in Israel, where an important population of cultured meat companies is present. In order to see which regulatory approval procedure is most robust and flexible, the proof of the pudding is in the eating. Sure thing however is that companies obtaining regulatory approval in Singapore will not want to limit their offerings to this small territory, but expand to others as well. In my view, mutual approval proceedings could beneficially influence each other, as data generated or approvals obtained in the one proceedings, could be used in the other. In terms of processing Novel Foods applications, I could only wish EFSA takes inspiration from the collaborative approach the SFA seems to take.
EFSA Guidelines bonanza: how to smoothly navigate the new transparency requirements?
Posted: March 26, 2021 Filed under: alternative protein, Authors, Food, novel food Comments Off on EFSA Guidelines bonanza: how to smoothly navigate the new transparency requirements? The world of food is changing at a rapid pace – alternative proteins becoming a new reality. Examples are products using microbial fermentation or cellular agriculture in their production process. If you are a company wanting to market such a product in the EU, there is a good chance you require a prior market authorization as a regulated product, such as a novel food (NF) or a GMO. In that case, you need to get up to speed with the following four new ESFA guidance documents that will all apply as of 27 March 2021.
The world of food is changing at a rapid pace – alternative proteins becoming a new reality. Examples are products using microbial fermentation or cellular agriculture in their production process. If you are a company wanting to market such a product in the EU, there is a good chance you require a prior market authorization as a regulated product, such as a novel food (NF) or a GMO. In that case, you need to get up to speed with the following four new ESFA guidance documents that will all apply as of 27 March 2021.
- Decision laying down the practical arrangements on pre-submission phase and public consultations
- Decision of EFSA’s executive director laying down practical arrangements concerning transparency and confidentiality
- Administrative guidance for the processing of applications for regulated products (update 2021)
- ESFA’s catalogue of support initiatives during the life-cycle of applications for regulated products (update 2021)
Document # 3 explains EFSA’s workflow in dealing with regulated products. It applies to applications for authorization of substances used in food and feed, food contact materials, NFs and GMO’s amongst others. The title of document # 4 is pretty self-explanatory. Examples of EFSA support initiatives range from general pre-submission and renewal advice to admin support to SMEs on applications and from EFSA guidance documents and scientific opinions to roundtables with industry associations.
Documents # 1 and 2 contain practical arrangements for companies that intend submitting scientific evidence regarding products requiring an EFSA evaluation. The full list of products is enumerated at the end of this post.* From a legal point of view, these guidelines implement the changes introduced in the General Food Law Regulation by the Transparency Regulation. We already covered the consequences of this Regulation in a previous blogpost, which in particular focuses on the relation between transparency and confidentiality.
This blogpost provides more detailed information on two topics representing a real change in the situation before 27 March, notably the general pre-submission advice and the notification of studies. To make this blogpost more concrete, it will target one specific category of regulated products within EFSA’s remit, notably NFs. For a reason. Currently, a great number of EU food companies active in the field of cultured meat are preparing their novel food applications. These companies need to be aware of these changes in the current regulatory framework.
General pre-submission advice (GPSA)
The GPSA to be provided by EFSA Staff concerns the rules applicable to and the content required for the NF application prior to its submission. It explicitly does not cover the design of the studies to be submitted. Aspects going beyond information available in any guidance documents are out of scope of pre-submission advice. Examples are questions regarding hypotheses to be tested or risk management. Furthermore, said staff shall not be involved in any preparatory scientific or technical work that is (in)directly of relevance for the NF application to be made. Also, the advice given shall be without prejudice and non-committal as to any subsequent assessment by EFSA (and the Member States via the PAFF Committee).
Prior to obtaining any GPSA, the applicant (and / or its representative) must register in the EFSA system supporting pre-submission activities and it must also request a pre-application ID. The pre-submission request can be introduced at any time before submitting a NF application. The European Commission nevertheless recommends submitting a request at least six months before introduction of the intended NF application.
The request for pre-submission advice shall be made in a dedicated form (“general pre-submission advice form”) made available by EFSA and a link should be made to the pre-application ID. Subsequently the applicant for an authorisation of a NF should provide all necessary information regarding the application, including a list of exhaustive questions. A maximum of two requests may be submitted under the same pre-application ID.
After a check that the questions asked are not out of scope, the applicant will be informed within 15 working days from the receipt of the advice form whether the submitted request is accepted or rejected. Companies expecting face-to-face meetings with EFSA need to know this will not happen automatically. Where possible, EFSA shall address the queries in writing. However, if EFSA considers that a discussion with the applicant might be useful to clarify certain aspects of the request, a meeting shall be organised. You then do not need to travel to Parma or elsewhere, as such meeting shall be conducted preferably by telco or webco. Only in exceptional cases a live meeting shall take place.
Any requests to be delt with in writing shall be answered within 15 working days after acceptance of the request. Any meeting to be set up shall be organised within 20 working days after acceptance of the request. EFSA shall draft a summary of the questions asked and not only send this to the applicant, but it will also publish it after the novel food application at stake has been considered admissible or valid.
Notification of studies
For transparency purposes, EFSA has established a database of studies commissioned or carried out by business operators to support any application requiring any type of scientific output, including a scientific opinion. The obligations to submit information on studies applies to both (A) potential applicants (i) having commissioned to a lab or external testing facility or (ii) carrying out such studies in in-house testing facilities and (B) to the labs or testing facilities carrying out such studies. Don’t be fooled by the wording “studies carried out” in relation to the notification required. In fact, timing is of the essence here, as all study notifications need to be submitted before the actual starting date of the study.
There may be justified justifications for not timely notifying studies. However, if EFSA does not consider the justifications provided by an applicant valid, the application needs to be re-submitted. As a consequence, the assessment of the validity of the application shall commence only six months after the notification of the studies according to the Transparency Regulation. The EFSA pre-submission guidance states the assessment of a re-submitted application shall commence 6 months after the re-submission of the application. This is not exactly the same, but it is clear that not complying with the studies notification requirement can be a serious showstopper, as a NF application supported by not-notified studies is not valid.
What exactly needs to be notified in the EFSA studies database? First of all, the study title, as well as an English translation thereof if the original title is not in English. Secondly, the applicant needs to be identified, after having first registered in the EFSA system for pre-submission activities. Thirdly the planned study starting and completion date, the laboratory where the study shall be carried out, as well as the study scope. In order to detail the study scope, the applicant should provide information on the intended study area (e.g. cultured meat), the study type (e.g. genotoxicity in vitro study) and the study objective (e.g. demonstrate that cultured meat does not pose any safety risks at the anticipated level of intake). The data entered can be modified, for instance if a study is delayed, the anticipated completion date can be aligned thereto. Information previously entered into the study database can also be withdrawn, but the applicant needs to provide a valid justification for that purpose. In the absence thereof, the same consequences will apply as when the applicant notifies its studies too late.
Once EFSA has received a valid or admissible application for a NF authorization, it shall publish this in order to launch the consultation of third parties on the application submitted. The rationale behind is that EFSA needs to have access to all relevant scientific data and studies available on the subject matter regarding the application at stake. Any communications on such consultation shall be published at EFSA’s website and shall remain open for three weeks, unless specified otherwise in sectoral Union law. For applicants, it is good to know that this public consultation in principle does not result in long delays. Upon closure of the public consultation, EFSA shall make publicly available all comments received. This will be done simultaneously with the publication of EFSA’s scientific output, which will address all relevant comments received.
Conclusion
What learnings can be drawn from the new EFSA Guidance documents? In the first place, that designing a safety evaluation strategy for a regulated product such as a NF has become even more important. By deciding as early stage as possible, what studies will be required to demonstrate the safety of your product, the chances that you can timely notify these studies in the EFSA study database shall increase. In this way, unnecessary delays in the appraisal of your application can be prevented. In the second place, it remains to be seen how helpful EFSA pre-submission advice will be, as it will be purely procedural and not material at all. At the same time, EFSA launched the new initiative of dedicated support for SME’s. Potentially, combination of the two reveals to be helpful.
—————————————————————————————————————————
*Products subject to transparency requirements and requiring an EFSA evaluation are the following:
- GMO’s (captured by both Directive 2001/18 and Regulation 1829/2003)
- Additives in animal nutrition
- Smoke flavouring used on food
- Food contact materials
- Food additives, enzymes and flavourings
- Plant protection materials
- Novel Foods
Labeling and Advertising of Alternative Protein
Posted: May 12, 2020 Filed under: alternative protein, Authors, Food, novel food Comments Off on Labeling and Advertising of Alternative Protein Sneak peek of Vitafoods Protein Summit
Sneak peek of Vitafoods Protein Summit
Consumers nowadays tend to include more and more plant-based products into their diet. For instance, a study ordered by DuPont Nutrition established double digits of growth for non-dairy ice-cream, dairy and cheese during the year preceding June 2018. More recently, NPR published an overview demonstrating which products were most consumed during the corona crisis. Amongst these, plant-based meat alternatives and oat milk were the biggest hits, demonstrating over 200 and 300 % growth respectively. Plant-based alternatives for dairy and meat will be discussed during the Protein Summit of the Vitafoods Conference in Geneva. As you may know, this conference was shifted from May to early September. In anticipation thereof, a webinar on Protein and Protein Alternatives took place on 12 May. During this webinar, I covered the labeling and advertising of these products. This blogpost offers a recap of my contribution thereto, targeting those who are interested in this topic but could not assist.
Regulatory requirements for market access
Obviously, there are other relevant aspects for alternative protein-based products than labeling and advertising, such as the regulatory requirements for market access. For some products, like those based on certain algae or on isolates from mung beans, most likely an authorization under the Novel Food Regulation will be required. This implies that the applicant will have to put together a product dossier demonstrating the safety of the product and submit this to the European Commission. This certainly applies for cultured meat products, unless they incorporate GMO steps in their production process and / or end product. In such case, market authorization will have to be obtained on the basis of the GMO Directive and the GMO Regulation. These topics will be dealt with during the Conference in September, detailing the scientific, economic and practical implications thereof.
Relevant general labelling rules
A number of labeling rules are of relevance for any food product, including those based on alternative protein. In fact, the cornerstone of labeling law, embodied in the Regulation on Food Information to Consumers, is the prevention of misleading. This can be done in various ways, but it is of the essence that at all times any confusion about the characteristics of a product is avoided. During the webinar, I brought up the example of Mylk to clarify this. Obviously, this is not a conventional dairy product, but a plant-based product. Do you think any confusion about the nature and / or the composition of this product could arise?
This should be decided based on the so-called Teekanne decision of the European Court of Justice (ECJ). According to this decision, it is prohibited to give the impression (by means of an image or a description) that a particular ingredient is present in a product, whereas this is not the case and the consumer can only find out when reading the list of ingredients. When this test is applied to the product Mylk, I am of the opinion it shall pass. Firstly, consumers will most likely not expect conventional milk, because of the twist in the product name. Secondly, FOP it states “dairy-free”. Thirdly, most plant-based dairy products are not stored in the fridge in the supermarket. Lastly, from the list of ingredients it is apparent this product is based on coconut cream.
Sector specific dairy legislation
For the dairy sector, important guidance was provided by the Tofutown decision by the ECJ. As detailed in an earlier blog post, we learnt from this decision that – in short – it is prohibited to use dairy names, such as “Tofubutter”, “Pflanzenkäse” and “Veggiecheese” for non-dairy products. This can be explained by the fact the dairy market is highly regulated, meaning that any specific dairy product has its own product standard. This standard should be met when manufacturing and marketing the product at stake. During the webinar, I mentioned the product standard for milk, defined as “normal mammary secretion obtained from one or more milkings without any addition thereto or extraction therefrom”. From this definition it follows why the use of the work milk in combination with a plant-based ingredient is, in principle, no longer allowed.
However, there are always exceptions to the rules, also in this case. These exceptions relate to so-called traditional use, like “coconut milk” (UK) and “lait d’amande” (France). Those exceptions are mentioned on a list drawn up by the European Commission. Furthermore, the word “milk” and designations used for milk products (e.g. cream, butter, yoghurt) can be used in association with one or more words designating certain composite products (famous example: “chocolate milk”). A condition precedent however is that milk is an essential part thereof, either in terms of quantity or for characterization of the product, and no constituent takes the place of milk.
Plant-based meat replacements
For these types of products, using (or not) the word “meat” is not so much an issue, because they are no conventional meat product. This issue is rather whether it is legally permitted to use certain meat product designations, such as “hamburger”, “sausage” and the like. In the US, we have seen so-called censorship bills in a great number of States. These are usually driven by the meat sector lobby, who fear unfair competition from their plant-based peers. There is fierce opposition against such censorship, amongst others from the Good Food Institute. Mid April, the GFI reported their lobby had been successful in Virginia, where the Governor vetoed label censorship.
In the EU, we have seen similar initiatives when the AGRI Committee of the European Parliament proposed a bill restricting the use of meat product names for meat alternatives. The status of this bill is yet undecided, as the current European Parliament that was inaugurated in 2019, did not yet vote on it. Fierce lobbying pro and con is however going on. We must therefore anticipate that if this bill turns into law, it will also result into restrictions of very popular terms. The alternatives for those popular terms are not so obvious yet: “lentil slices?” or “carrot tubes?”.
Please bear in mind that restrictions can also stem from national Member State laws based on reserved product designations. In the NL for instance, the name “minced meat” and “tartar” are such reserved product designations that can only be used for products meeting exactly the relevant legal specifications.
Comparative advertising
When discussing the advantages of new over conventional protein products, certain advertising standards should be taken into account. In the EU, one of the ways to avoid misleading regarding your alternative protein product is to not emphasize certain characteristics that your product does not have. In this context, I discussed in the webinar a commercial that was made for BECEL margarine (FLORA in UK) that was shown both in the Netherlands and in Belgium. The text stated “Plants are the new cows. They are outside in the field whole year long. They provide seeds, that are a source of omega-3, which is good for your heart. BECEL is 100 % plant-based and good for your heart.”
According to a complaint filed with the Dutch Advertising Code Committee (similar to Advertising Standards Authority in UK), the comparison made between plants and cows was misleading. Rationale: it was suggested that plants had a more positive effect on the environment than dairy products made from milk. The complaint was dismissed. The ACC considered that the commercial did not at all compare the advantages of plant-based products to the disadvantages of conventional dairy. In fact, it only stressed the positive health effects of BECEL, due to its plant-based ingredients. The commercial appeared an effective (and funny!) way of advertising alternative dairy products.
Takeaway
Alternative protein products are food products like any other, so make sure that when marketing these, you are up to speed with all applicable general labeling requirements. Furthermore, take into account any sector specific standards, like the ones that apply for dairy products. Also, please note that at Member State level, further restrictions on the use of particular product names may apply. Finally, when advertising these products, make sure to avoid any misleading and know the rules for comparative advertising. This will be of particular relevance, especially once further labeling standards will evolve at EU level as initiated by the AGRI Committee of the European Parliament. Stay tuned – we will.
Copyright image: Nanne Meulendijks – please contact the artist for any further use.
Trends from Silicon Valley for meat alternatives
Posted: October 2, 2019 Filed under: Authors, clean meat, Food, novel food Comments Off on Trends from Silicon Valley for meat alternatives What does the future of our meat look like? This topic was the subject of the Annual Conference of the Good Food Institute (GFI), which took place in San Francisco last month. GFI is an American non-profit organization that works with entrepreneurs, scientists and investors to make groundbreaking good food a reality. GFI focuses on the development of alternatives to animal products, consisting of plant-based products and products manufactured using cellular techniques (“cultured meat” or “cultured fish”).
What does the future of our meat look like? This topic was the subject of the Annual Conference of the Good Food Institute (GFI), which took place in San Francisco last month. GFI is an American non-profit organization that works with entrepreneurs, scientists and investors to make groundbreaking good food a reality. GFI focuses on the development of alternatives to animal products, consisting of plant-based products and products manufactured using cellular techniques (“cultured meat” or “cultured fish”).
Trends from Silicon Valley also in EU
The issues raised at this conference are also relevant to Europe. In the Netherlands, for example, the Nutrition Centre advises that it is better for human health to have a more plant-based diet than has been the case until now. For example, the risk of cardiovascular disease would decrease if the diet included less meat and more wholegrain cereal products, legumes, vegetables, fruit and vegetable meat substitutes.
GFI sessions: trending topics
In the country where the Beyond Meat citizen comes from, it was recognized that a large-scale breakthrough of meat alternatives has not yet taken place. The titles of the presentations were telling: “Marketing to Meat Eaters: How to Reach the Other 95 Percent of American Consumers“, “Capitalizing on Change: How Investors Accelerate the Plant-Based and Cell-Based Industries” and “Addressing the Key Challenges in Commercializing Cell-Based Meat“. Participants in the last session of the conference (“Cell-Based Meat Entrepreneurship“) were JUST and Memphis Meats, the companies that are said to be “closest to the market” with their cultured meat products. According to JUST, they will launch a cultured meat burger still this year. But that’s what they say every year.
Plant-based meat substitutes in the US
What is legally required to be able to market meat alternatives? Of course, it all starts with the name: what can you call these products? The basic rule, both in the US and in the EU, is that consumers must not be misled. In the case of plant-based products, this principle is applied in various ways in the United States. For example, the burger of Beyond Meat (which is now also available in the Netherlands) is simply referred to as the BEYOND BURGER and described as “the world’s first plant-based citizen that looks, cooks, and satisfies like beef…“. Impossible Foods also refers to its company name in its IMPOSSIBLE BURGER. This product is not yet on the market in the EU, partly because it contains a protein of which the regulatory status in the EU is not entirely clear (so-called leghomoglobin). This protein is obtained on the basis of fermented yeast, possibly using GMO techniques. Finally, Morning Star (“It easy eating green“), when selling its plant-based burgers, refers to the flavour-defining ingredient that they contain, for example “mediterranean chickpea burgers” and “spicy black bean burgers“. Confusion with conventional meat products does not seem to be an issue here.
And what about the EU? Famous example of the Vegetarian Butcher
In the meantime, more and more plant-based meat substitutes are also being sold in the EU. One of the best-known examples of (alleged) confusion about product names in the Netherlands concerned the case of the Vegetarian Butcher, a company that was acquired by Unilever in December 2018. At the end of 2017, the Dutch Food Safety Authority (NVWA) asked this company to modify the labels of the products offering a substitute for tuna, chicken and bacon (all spelled with a twist in Dutch), because these would be misleading. It turned out afterwards that the criticism concerned only the information on the website. The NVWA apologised and the Vegetarian Butcher did not have to change any product labels.
Proposal for a ban on Vegaburger in the European Parliament
The next shockwave took place at European level when, in April of this year, the Committee on Agriculture and Rural Development (AGRI Committee) of the previous European Parliament adopted a proposal to ban the use of meat names for plant-based products. On the basis of this proposal, it would no longer be possible to use the names ‘steak’, ‘sausage’, ‘ham’, etc. for plant-based products. A new European Parliament has now been elected. It has been reported from Brussels sources that the new AGRI Committee has taken the old proposal as a starting point for a working document. However, so many forces are working together against the previously envisaged ban that the chances of this happening are not considered to be very great. Arguments against this ban are that it would inhibit innovation in the EU, harm consumer rights and simply not be necessary: consumers are not so easily confused about plant-based meat substitutes. With the slogan “Stop the vegaburger ban“, Pro Veg launched a petition against this ban.
Cultivated meat in the US
Currently, about 30 companies active in the field of cultured meat worldwide. The companies that are most advanced with their innovations are located in the US. Menphis Meat, for example, focuses on the development of beef and poultry from the lab. This company has received investments from both well-known names (Bill Gates, Richard Branson) and from the “classic” meat industry (Tyson and Cargill). JUST focuses on the development of, among other things, chicken meat from the lab. The company has already launched an egg-free mayonnaise, that was the object of discussions with the FDA. According to the current product standard for mayonnaise, this product must contain eggs, otherwise it cannot be called “mayonnaise”. This problem was solved by changing the name of the product (“JUST Mayo”). How cultured meat will be named upon its market introduction is yet an open question. However, five companies have joined forces: they form the so-called Alliance for Meat Poultry and Seafood Innovation to approach the supervisory authorities with one voice. GFI has also developed a narrative framework to familiarise consumers with this technology and it published the results of market research into favorite names. For the time being, “cultivated meat” and “cultured meat” are at the top in terms of “appeal” and accuracy.
Cultured meat in the EU
In the EU, the question remains whether cultured meat can actually be called ‘meat’ at all. The number of arguments for and against is more or less balanced. We reported earlier on this topic in our blogs How do we get cellular ag products to the market and Regulatory pathways for clean meat in the EU and the US; not much progress has been made since that time. The main argument in favor is that when a product is identical to conventional meat at a molecular and nutritional level, producers want to be able to use this name as well. In addition, the use of the term ‘meat’ is very functional in terms of consumer orientation. Furthermore, there do not seem to be any direct legal objections to the use of ‘meat’ for cultured meat, since the Agricultural Products Standards Regulation does not contain a specific product standard for meat, unlike, for example, dairy products. Arguments against the use of ‘meat’ are based, among other things, on the Hygiene Regulation. This Regulation defines ‘meat’ as certain parts of a variety of animals, including bovine and porcine animals and poultry. It is questionable whether a product obtained from a single cell derived from one of these animals complies with this definition. Furthermore, the Hygiene Regulation refers in various places to the slaughter of farmed animals. Of course, slaughter does not apply to cultured meat – that’s the whole point. Finally, certain meat names are subject to specific regulations. In the Netherlands for example, the name ‘tartar’ may only be used for minced meat from cattle with a maximum fat content of 10 % based on specific Commodities Act legislation.
Conclusion
The future of our meat is taking shape. Plant-based meat substitutes are already on the market in large numbers and these products are getting more and more shelf space in the supermarket. The challenge for these products is to keep that shelf space. A certain amount of time will elapse before cultured meat will be available on a large scale on the shelf. It is expected that the first company will submit a request for Novel Food authorisation sometime next year. It will take about 18 – 24 months to complete such an authorisation procedure. By then we will have reached 2022. This may seem a long way off for the consumer, but the preparation by the producer has been a reality for a long time. Also, it was learned via the grapevine that EU-based cultured meat companies are getting organized. Watch that space!
Transparency vs. Confidentiality: EFSA’s new role as from March 2021
Posted: July 31, 2019 Filed under: Authors, Food, Information, novel food Comments Off on Transparency vs. Confidentiality: EFSA’s new role as from March 2021 Last month, the European Council formally adopted the new Regulation on the transparency of the EU risk assessment in the food chain, which will be applicable as of 26 March 2021. As is in the name, the new provisions aim at increased transparency of EU risk assessment, which for a large deal means strengthening the reliability, objectivity and independence of the studies used by European Food Safety Authority (“EFSA”). As EFSA has been accused more than once of conflicts of interest, this is, in principle, a welcome development. For FBOs, the implications are however important – as will be demonstrated below.
Last month, the European Council formally adopted the new Regulation on the transparency of the EU risk assessment in the food chain, which will be applicable as of 26 March 2021. As is in the name, the new provisions aim at increased transparency of EU risk assessment, which for a large deal means strengthening the reliability, objectivity and independence of the studies used by European Food Safety Authority (“EFSA”). As EFSA has been accused more than once of conflicts of interest, this is, in principle, a welcome development. For FBOs, the implications are however important – as will be demonstrated below.
The upcoming changes follow from the Commission Communication on the European Citizens Initiative on glyphosate, in which initiative EU citizens called for more transparency in scientific assessments and decision-making, and build upon the findings of the fitness check of the General Food Law. The new provisions will mainly amend the General Food Law. For reasons of consistency, the new provisions also introduce changes to eight legislative acts dealing with specific sectors of the food chain, including the Novel Food Regulation, the Additives Regulation and the GMO Regulation. Based on the publicly available proposal for the meanwhile adopted provisions on the transparency of EU risk assessment, this blogpost provides an overview of what applicants will be facing in authorization procedures for innovative food products in terms of transparency and confidentiality.
Pre-submission phase: union register of commissioned studies and general advice
Before submitting an authorization application, for instance for a Novel Food like cultured meat or insects or for a new additive, applicants will have to notify EFSA of any study they commissioned to support a future authorization application. These studies will become part of the union register of commissioned studies as managed by EFSA. The submitted studies will be made public only when an actual application follows and insofar the studies do not contain confidential information, regarding which a request for confidential treatment has been granted. The idea behind the union register is that companies (potentially) applying for an authorization submit all related information, which subsequently allows EFSA to cross-check the information on the studies performed. This way, applicants can no longer hold back unfavorable studies. This notification obligation also applies to laboratories in the EU that carry out those studies, but obviously not to non-EU laboratories as they fall outside the scope of new provisions. According to the EC’s fact sheet of 13 June 2019, consequences of non-compliance with the notification obligation shall result in a negative temporary stop in the risk assessment.
At the same time, applicants have the right to request EFSA for advice on the relevant provisions and the required content of the application for authorization in the pre-submission phase. This procedure is a response to industry demand, especially of SMEs, for further support in the preparation of applications. The advice shall, however, be provided without the input of the Scientific Panels that are in charge of the actual scientific assessment and shall not cover the specific design of a study. Furthermore, EFSA’s advice shall be made public. Based on the publicly available proposal for the transparency provisions, it seems that this will be done before an actual authorization application is being submitted.
Submission phase: citizens’ access to studies vs. confidentiality
 The default setting under the new provisions is that all scientific data, studies and other information supporting applications for authorizations shall be made public by EFSA. While the new provisions aim at ensuring that stakeholders and the public have access to key safety related information being assessed by EFSA, duly justified confidentiality information shall not be made public. The new rules specify which types of information may be considered as such. As listed in the publicly available proposal, this includes production methods, the quantitative composition of the substance or product at stake, certain commercial relationships and other sensitive business information. It is the responsibility of the applicant to demonstrate that making public the information concerned would significantly harms the (commercial) interests concerned. Only in very limited and exceptional circumstances relating to foreseeable health effects and urgent needs to protect human health, animal health or the environment, confidential information shall be disclosed.
The default setting under the new provisions is that all scientific data, studies and other information supporting applications for authorizations shall be made public by EFSA. While the new provisions aim at ensuring that stakeholders and the public have access to key safety related information being assessed by EFSA, duly justified confidentiality information shall not be made public. The new rules specify which types of information may be considered as such. As listed in the publicly available proposal, this includes production methods, the quantitative composition of the substance or product at stake, certain commercial relationships and other sensitive business information. It is the responsibility of the applicant to demonstrate that making public the information concerned would significantly harms the (commercial) interests concerned. Only in very limited and exceptional circumstances relating to foreseeable health effects and urgent needs to protect human health, animal health or the environment, confidential information shall be disclosed.
Specific sectoral food legislation will also include a list of certain types of information that may be treated confidentially. For example, a proposed change to the GMO Regulation includes the specification that certain DNA sequence information and breeding patterns and strategies may be treated confidentially. Next to the confidentiality rules, existing intellectual property rights, data protection rights and data exclusivity provisions for proprietary data, remain applicable.
When an applicant submits a request for confidentiality, it shall 
provide a non-confidential version and a confidential version of the submitted information. If and when EFSA receives the submitted information, for example in case the Commission requires a scientific opinion as part of a novel food procedure, it shall publish the non-confidential version without delay and shall decide on the requested confidentiality within 10 days. Insofar the confidentiality request has not been accepted, EFSA shall subsequently publish the additional information after 2 weeks from the notification of its decision. Applicants that do not agree with EFSA’s decision may, under certain circumstances, challenge the decision before the Court of Justice of the European Union.
Once EFSA has published all the non-confidential information and before it carries out its scientific assessment, EFSA may consult third parties, including citizens, to identify whether other relevant information is available on the substance at stake. This new provision serves to mitigate the public concerns that EFSA’s assessment is primarily based on industry studies, more in particular on studies ordered by the applicant. The results of these consultations shall also be made public, as will be minority opinions to EFSA’s scientific output.
Renewals
In case of a request for the renewal of an authorization, the applicant of the renewal will have the obligation to inform EFSA on its planned studies under the new rules. EFSA shall subsequently consult stakeholders and the public on the planned studies and, taking into account the received comments, provide the applicant with advice on the content of the intended renewal application. The reason behind this pre-submission procedure is to make use of the existing experience and knowledge on the substance or product in question. The European Commission expects that the notification obligation will have a positive effect on the evidence base of EFSA, avoids the unnecessary repetition of studies, and will provide the potential applicant with useful advice. It should, however, be noted that also in this case EFSA’s advice will not stand in the way of the subsequent assessment of the renewal application by the Scientific Panels.
Additional controls on the conduct of studies
The new provisions also include measures to ensure the quality and objectivity of the studies used by EFSA for its risk assessment. First of all, the European Commission will have the right to perform controls, including audits, to verify the compliance of testing facilities with relevant standards. Competent authorities of the Member States will be involved in these controls. Through coordination with OECD GLP auditing programs, the control in non-EU countries will be facilitated as well. Secondly, the European Commission will have the right to request EFSA to commission verification studies. Such action shall only be taken in exceptional circumstances, such as in case of serious controversies or conflicting results, and will be financed by the EU.
Conclusion
The new provisions address public concerns regarding transparency, while on the other hand acknowledge that confidentiality is key to not cut down on incentives for innovation by food businesses. In other words: the new provisions seek to balance transparency and confidentiality. Food businesses are advised to substantively consider their commercial interests when submitting a dossier for authorization, since confidentiality will only be granted upon duly justified requests. Moreover, the union register for submitted studies forces food businesses to carefully plan their studies at an early stage since also studies with less favorable outcomes will in principle be made public and scrutinized on inconsistencies with later studies. At the same time, more guidance is expected from EFSA in the pre-submission phase, possibly speeding up the actual authorization procedure. Given the fact that the new provisions will apply from less than 20 months from now, food businesses may consider speeding up or delaying their application for authorization, depending on their needs.
Regulatory pathways for clean meat in the EU and the US – differences & analogies
Posted: March 15, 2019 Filed under: Authors, clean meat, Food, novel food Comments Off on Regulatory pathways for clean meat in the EU and the US – differences & analogies Now that the US regulatory framework is shaping up, the analogies and differences with the European regulatory framework for market access for clean meat becomes more and more clear. This blogpost reports these analogies and differences, based on the agreement that the FDA and USDA recently concluded on their cooperation in the field of clean meat.
Now that the US regulatory framework is shaping up, the analogies and differences with the European regulatory framework for market access for clean meat becomes more and more clear. This blogpost reports these analogies and differences, based on the agreement that the FDA and USDA recently concluded on their cooperation in the field of clean meat.
Joint action FDA-USDA in regulating cell-based meat
On 7 March 2019, the FDA and the USDA concluded a formal agreement (“7 March Agreement”) on their cooperation to oversee the production of cell-based meat from livestock and poultry. As mentioned in an earlier blogpost, in the EU the European Commission is the one stop shop for obtaining a Novel Foods authorization. In the US however, foods can be subject to regulatory oversight by multiple federal and state agencies. In general, USDA regulates meat and poultry, including the inspection of establishments that slaughter such animals or otherwise process meat and poultry products. FDA generally regulates all other food, including fish and certain other meat and poultry products, such as bison, rabbits, and wild turkeys and ducks.
Essence of the 7 March Agreement
The 7 March Agreement is quite a relief for the market players concerned. For instance, the Good Food Institute issued a press release, stating “The agreement is a significant step forward in providing a transparent and predictable regulatory path to market for cell-based meat, which will help to ensure that the U.S. does not fall behind Israel, China, Japan, the Netherlands, Singapore, and other countries that are moving quickly to ensure a clear path to market for this method of meat production”. In essence, according to the 7 March Agreement, FDA is competent to oversee cell culture and production up to harvest of the cultured meat cells, whereas USDA takes over responsibility as of harvest up to and including the actual production of clean meat products. For clarity, clean fish is not included in the 7 March Agreement, as this under the remit of FDA. It seems safe to assume however that what the agencies will do for clean meat and poultry, they will do for clean fish. This assumption is based on the agreement that the agencies will develop joint principles for product labeling to ensure that products are labeled consistently and transparently.
Analogies between EU and US
By far the most important analogy is that both regulatory systems aim to assure that clean meat products hitting the market are “safe, wholesome and unadulterated” (see point 4 B (3) of 7 March Agreement). In fact, to the EU Novel Foods Regulation aims to ensure “the effective functioning of the internal market while providing a high level of protection of human health and consumers’ interest.” In order to achieve this purpose, both regulatory systems require prior market authorization, but the way such authorization procedure is put in place is in fact quite different.
Differences between the EU and US
In order to visualize the differences between the two regulatory systems at each side of the Atlantic, I have created the schematic overview below. On the left side, I summarized the authority of the FDA and the USDA under the 7 March Agreement. On the right side, I outlined how legal authority is attributed in the EU and in its Member States. As a reference Member State, the Netherlands (“NL”) has been retained, as this is one of the EU countries where clean meat activities are prominent.
| FDA (“pre-harvest”) | EU | |
| 1. | Pre-market consultation to evaluate production materials & processes | No formal pre-market consultation procedure in EU Novel Foods framework, except the optional consultation at Member State level in case of doubt whether the product qualifies as a Novel Food (which is clear in the case of clean meat) |
| 2. | Oversee cell collection and quality of cell banks | Oversight of preparatory production steps, as well as registration of a company as a food business operator (FBO) will be done at Member State level. In the NL, FBOs working with products from animal origin, require a so-called recognition (“erkenning“). This is a more detailed procedure (average term: 8 weeks) than the mere registration of a FBO (average term: few days). |
| 3. | Oversee production process until harvest | |
| 4. | Ensure companies comply with FDA requirements: facility registration, cGMP and other applicable food legislation | |
| 5. | Where needed: issuing regulations or guidance or additional requirements re. # (2) and (3) to ensure that biological materials exiting the culturing process are safe (FFDCA) | EU Hygiene regulation targeting food of animal origin (853/2004) to apply and potentially national legislation as well. In the NL, the Commodities Act Decrees on hygiene and on the preparation and packaging of foodstuffs are applicable. Additional requirements (“conditions of use”) may also be included in individual Novel Food authorizations. |
| 6. | Inspections and enforcement directed at safety of cell banks and culturing facilities | Inspections and enforcement are done at Member State level. In the NL the responsible entity is the Dutch Food Safety Authority. |
| USDA (“post harvest”) | ||
| 7. | Determine whether harvested cells are eligible to be processed in meat or poultry products | The NF-framework requires FBOs in their application for market authorization to specify the source of the product, its production process and typical compositional features. No additional eligibility test upon cell harvest prior to production of food products. |
| 8. | Require each clean meat company to obtain a so-called grant of inspection | Not required under EU legal framework. Registration (or recognition) with the competent Food Safety Authority provides the authority with the legal basis for inspection. Obviously, a Novel Food authorization must also be obtained before placing the product on the market. |
| 9. | Conduct inspections in establishments where cells cultured from livestock and poultry are harvested, processed, packaged or labeled to ensure that the resulting products are safe, unadulterated, wholesome and properly labeled. | Inspections will be executed at a Member State level, based on the Official Controls Regulation 854/2004 targeting products of animal origin for human consumption inter alia. |
| 10. | Pre-approval of labeling of clean meat products and inspection thereof | No pre-approval of product labels under EU NF-framework. It is the responsibility of the FBO himself to comply with applicable labeling legislation, such as the Food Information to Consumers Regulation 1169/2011. |
| 11. | Where needed: develop additional requirements to ensure the safety and accurate labeling of clean meat products | Safety and Labeling provisions already in place at EU level. These are embodied in the General Food Law Regulation 178/2002 and the Food Information to Consumers Regulation 1169/2011 respectively. Furthermore, specific labeling requirements may be included in Novel Food authorizations. Also, post-market monitoring requirements may be imposed. In any event, FBOs should inform the Commission of any new relevant information regarding the safety of the NF they have placed on the market. |
| 12. | Enforcement actions re. adulterated or misbranded food products | See comment to # 6. Competitors, consumers and watchdog organizations may also bring cases re. misleading food information before self-regulatory bodies. From example, unpermitted references to “meat” could be a topic of such cases. |
| Both FDA and USDA | ||
| 13. | Each entity to cooperate with the other upon transfer or regulatory oversight at harvest | |
| 14. | Each party to notify the other if it identifies objectionable conditions resulting in adulterated / misbranded clean meat products |
No pre-market consultation in the EU
One of the most striking differences is that under the EU Novel Foods procedure, there is no formal pre-market consultation procedure to evaluate production materials and processes. I consider this to be a flaw in the law. Under the former Novel Foods Regulation that was in place until 1 January 2018, applications had to be filed with the competent authority at a Member State level. These authorities, at least in the Netherlands, were often willing to answer questions that arose during the preparation of applications. Under the current Novel Foods Regulation, applications have to be directly filed with the European Commission via an e-portal. There are no formal procedures to contact either the European Commission, or EFSA to answer questions on applications. If you manage to have informal consultation with an EFSA representative, no questions regarding particular applications will be answered, so a sectoral approach is your best bet. In short, we will have to learn by doing!
Written EFSA guidance
Where guidance by the Commission or EFSA on a personal level is not available, there is ample guidance based on a 2016 EFSA Scientific Opinion and a 2018 EFSA Technical Report. In fact, the 2016 Opinion provides detailed comments as to the various data to be provided in the Novel Food application, for instance on the identity of the Novel Food. For clean meat in particular, the identity of the cells from which the meat product is cultured should be provided, as well as the cell substrate used during the cultivation process, which should also be described in detail. The 2018 Report provides applicants with a completeness checklist, covering all items that should be specified in a Novel Food application. It also provides a helpful overview table of study reports contained in the technical dossier.
Enforcement
Contrary to the US, where enforcement is done by the authorities who also define the regulatory framework, enforcement in the EU is a national affair. This means that not the European Commission, nor EFSA are involved, but the national competent authorities. In the Netherlands, this is the Dutch Food Safety Authority, whose enforcement policy is on public record. For instance, enforcement measures directed at marketing clean meat without a proper Novel Food authorization translate into a penalty and the prohibition to further market the product. Since enforcement is a national affair, measures may vary from Member State to Member state. When marketing clean meat in Europe, it is therefore not sufficient to be familiar with the EU framework but one is strongly recommended to also gain local advice.
Open ends
Open ends still occur at both sides of the Atlantic. In the US, the 7 March Agreement has given a flavor on how the FDA and USDA will cooperate, but “it does not create binding, enforceable obligations against either Agency.” So, it needs to be seen how this will work out in practice, for instance regarding the pre-approval of labels. Will the authority at Federal level take away the necessity for evaluation at State level? In the EU, it remains to be seen what type of studies EFSA will want to see to specify nutritional and toxicological information, as well as allergenicity: literature studies or rather in vitro or in vivo data?
At FoodHealthLegal, we will closely monitor the regulatory developments in the clean meat space, so stay tuned!
Credits for graphics: Mosa Meat
Cannabis derived food products – what’s the current state of play?
Posted: February 25, 2019 Filed under: Authors, cannabidiol, cannabis, Enforcement, Food, Health claims, novel food Comments Off on Cannabis derived food products – what’s the current state of play? Recently, CBD food products were qualified as Novel Foods requiring a market authorization. The lively trade in these products therefore currently seems to be at risk. However, not all cannabis derived products are Novel Foods. What is the current state of play regarding these products and how is enforcement going to look like?
Recently, CBD food products were qualified as Novel Foods requiring a market authorization. The lively trade in these products therefore currently seems to be at risk. However, not all cannabis derived products are Novel Foods. What is the current state of play regarding these products and how is enforcement going to look like?
Current state of play re. cannabis derived products
In the European Union, the cultivation of Cannabis sativa L. varieties is permitted provided they are registered in the EU’s ‘Common Catalogue of Varieties of Agricultural Plant Species’ and the tetrahydrocannabinol (THC) content does not exceed 0.2 % weight per weight. The Common Catalogue is embodied in the EC Plant Variety database, which currently lists 68 species of Cannabis sativa. Some products derived from the Cannabis sativa plant or plant parts such as seeds, seed oil, hemp seed flour and defatted hemp seed have a history of consumption in the EU and therefore, in principle, are not novel.
What’s new?
This is different for extracts derived from Cannabis sativa L. and derived products containing cannabinoids, such as cannabidiol (CBD). It follows from a recent clarification of the Novel Food Catalogue that these products are considered Novel Foods, as a history of consumption regarding these products has not been demonstrated. This applies to both the extracts themselves and any products to which they are added as an ingredient. If for instance CBD is added to hemp seed oil, the product can no longer be marketed just like that and requires market authorization. The status of Novel Food also applies to extracts of other plants containing cannabinoids and to synthetically obtained cannabinoids.
How does the market of CBD food products currently look like?
Currently, the market in CBD food products is flourishing. A variety of CBD nutraceutical products is being offered for sale, such as HempFlax CBD, CBD oil, but also CBD-infused tea, honey or sweets. Although there is no hard-scientific evidence, many health benefits are connected to CBD food products, such as stress reduction, good night rest and providing energy and increasing resistance. Contrary to products containing THC (tetrahydrocannabinol), which is also extracted from cannabis, you do not get high on CBD food products, as this is not a psychoactive substance.
Medicinal use of cannabis
The use of cannabis derived CBD in food is not to be confused with medicinal use of cannabis. In most cases of medicinally applied cannabis, the active ingredient is THC, not just CBD, or a combination of THC and CBD. Although medicinally applied cannabis does not play a role in the cure of diseases, scientific publications show it can alleviate suffering from diseases, for instance nausea, decreased appetite, slimming or weakening due to cancer.
Consequences for business of the change in legal framework
Due to the qualification of CBD food products as Novel Foods, the lively trade in these products is currently at risk. Any Novel Food has to obtain a market authorization in order to get market access. CBD food products currently marketed may face enforcement measures, unless they can benefit from the transition regime laid down in the Novel Foods Regulation. According to this transition regime, any product that did not fall within the scope of the former Novel Foods Regulation, was lawfully marketed prior to 1 January 2018 and for which an application for market authorization is filed before 2 January 2020, can continue to be marketed until an authorization decision has been taken. While this transition period is in principle drafted for Novel Foods that fall into one of the new novel food categories under the new Novel Foods Regulation, it is in the spirit of the transition regime to also include the CBD scenario.
Pending CBD-application and expected EFSA opinion
Currently, one application for the authorization of a CBD food supplement is pending. The application was made by the company Cannabis Pharma from the Czech Republic and is based on publicly available safety and toxicological information and toxicity reviews. More in particular, the scientific data has been gathered from acute and long-term toxicity studies in animals and tolerance studies in humans. The data package submitted aims to support the safety of the use of CBD in food supplements for adults with a daily intake of up to 130 mg or 1.86 mg/kg body weight. It is reported by various sources that an EFSA opinion is awaited this March (see here and here).
Any benefits for the CBD market of a positive EFSA opinion?
Contrary to the situation under the former Novel Foods Regulation, the authorizations granted under the current Regulation have a generic nature. This means that any other company meeting the conditions of use stated in the authorization, would be at liberty to market CBD food supplements as well. The pending application made by Cannabis Pharma is therefore followed with great interest by the CBD market. As they do not seem to rely on data protection, a granted authorization would pave the way for other food supplement companies. It is not certain if this will happen still this year. If and when EFSA grants a positive opinion this March, the European Commission still has 7 months to submit an implementing act to the PAFF Committee. Upon a positive opinion of the PAFF committee, such implementing act could be quickly adopted. If the PAFF Committee has no opinion or a negative opinion, 1 or 2 months should be added to the procedure as a minimum.
Ireland: some CBD food products can be marketed
Meanwhile, there is some guidance available at Member State level. The Irish Food Safety Authority notes that recently a large number of CBD food products entered the market, typically marketed as food supplements in liquid or capsule form. Depending on the manufacturing process applied, the trade in CBD oil is not prohibited, as this oil naturally contains low levels of CBD, which is considered a non-psychoactive compound. This applies to CDB oil obtained by cold-pressing the hemp seeds. If and when the oil is obtained by supercritical CO2 extraction, then a Novel Food authorization is mandatory.
Denmark: available guidance not crystal clear
According to the Danish Ministry of Environment and Food, a number of Cannabis-derived products are not considered Novel Food, notably hemp seeds, seed flour, protein powder from seeds and seed oil from the Cannabis sativa L. varieties listed in the EC Plant Variety Database that are free from or contain low levels of THC. If these products contain CBD, the regulatory status is not exactly clear. According to the guidance of the Danish Food Ministry, the current status is that pure cannabidiol as well as hemp products with high (concentrated) levels of CBD or other cannabinoids are covered by the Novel Foods Regulation. It is not explained what is understood by “high levels of CBD”, but on the other hand an absolute prohibition to market these products in Denmark does not seem to apply.
Absolute prohibitions: Belgium and Austria
Other Member States seem to be stricter than Ireland or Denmark. For instance, the Austrian Health Ministry has made it perfectly clear that food products containing any type of cannabinoid extract without a Novel Food authorization are prohibited to be put on the market. In Belgium, the Federal Agency on Safety in the Food Chain has clarified that the production and marketing of food products based on cannabis is prohibited. The rationale is that the plant Cannabis sativa is mentioned in an annex to a national Decree listing dangerous plants that cannot be used for food production. The prohibition primarily seems to target the potentially dangerous substance of THC and allows derogations on a case-by-case basis, but not regarding food products containing CBD. These are considered Novel Foods requiring a market authorization.
Enforcement directed against (medical) claims in the Netherlands
Until CBD was declared a Novel Food, the trade in CBD food products was not prohibited in the Netherlands. Contrary to the substance TCH, the substance CBD is not mentioned in the Dutch Opium Act, listing prohibited substances having a psycho active effect. This does not mean that the trade in CBD food products was allowed just like that. In practice, enforcement in the Netherlands has been directed against the use of any unauthorized medical claims. A medical claim is any information according to which a food product could have a therapeutic or prophylactic effect. When using such a claim, one comes into the realm of the Medicinal Product Act, according to which it is prohibited to market and advertise any medicinal product without a market authorization. The Dutch Food Safety Authority announced fines up to
€ 10.000 regarding the sale of CBD food products in several cases (see here and here). Any food business operator that is serious about his business in CBD food products will therefore not only check the applicability of the Novel Foods Regulation to his products, but also carefully draft his advertisement for this type of product.
Conclusion
The production and marketing of food products derived from Cannabis sativa L. in the EU has been considerably restricted since CDB food products were recently declared to be Novel Foods. However, not all cannabis-derived food products require market authorization. Pending the evaluation of the Novel Food application filed for a CBD food supplement by the Czech company Cannabis Pharma, it is worthwhile for other CBD food products to verify whether they can benefit from the so-called transition regime embodied in the Novel Foods Regulation. Due to differences between legislation in the Member States, this may differ from country to country. Also, it is important to carefully position your CBD food product, in order to avoid any medical claims.
The author ackowledges Jasmin Buijs, paralegal at Axon, and Max Luijkx, intern at Axon, for their valuable input.